Download to read offline




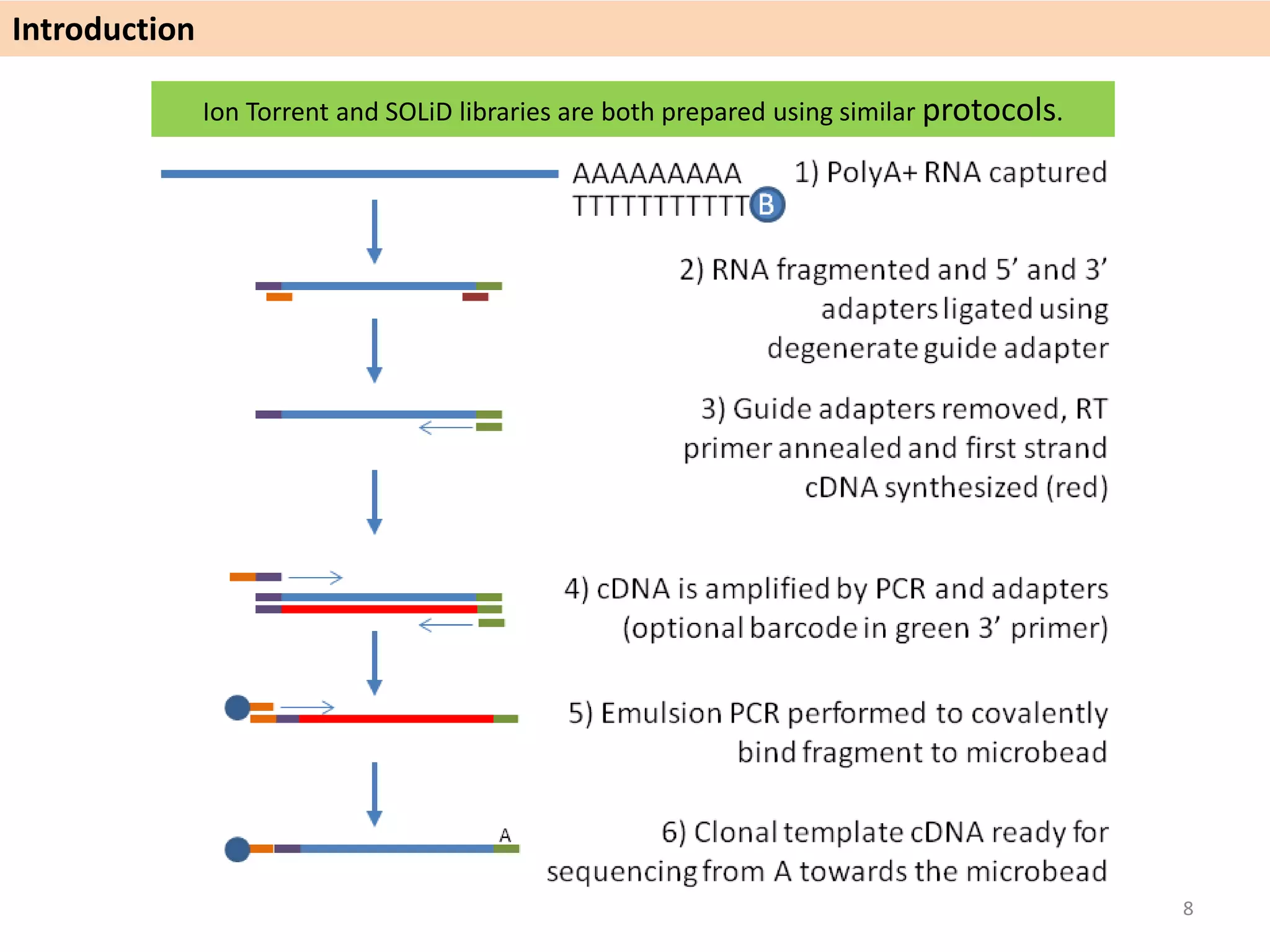
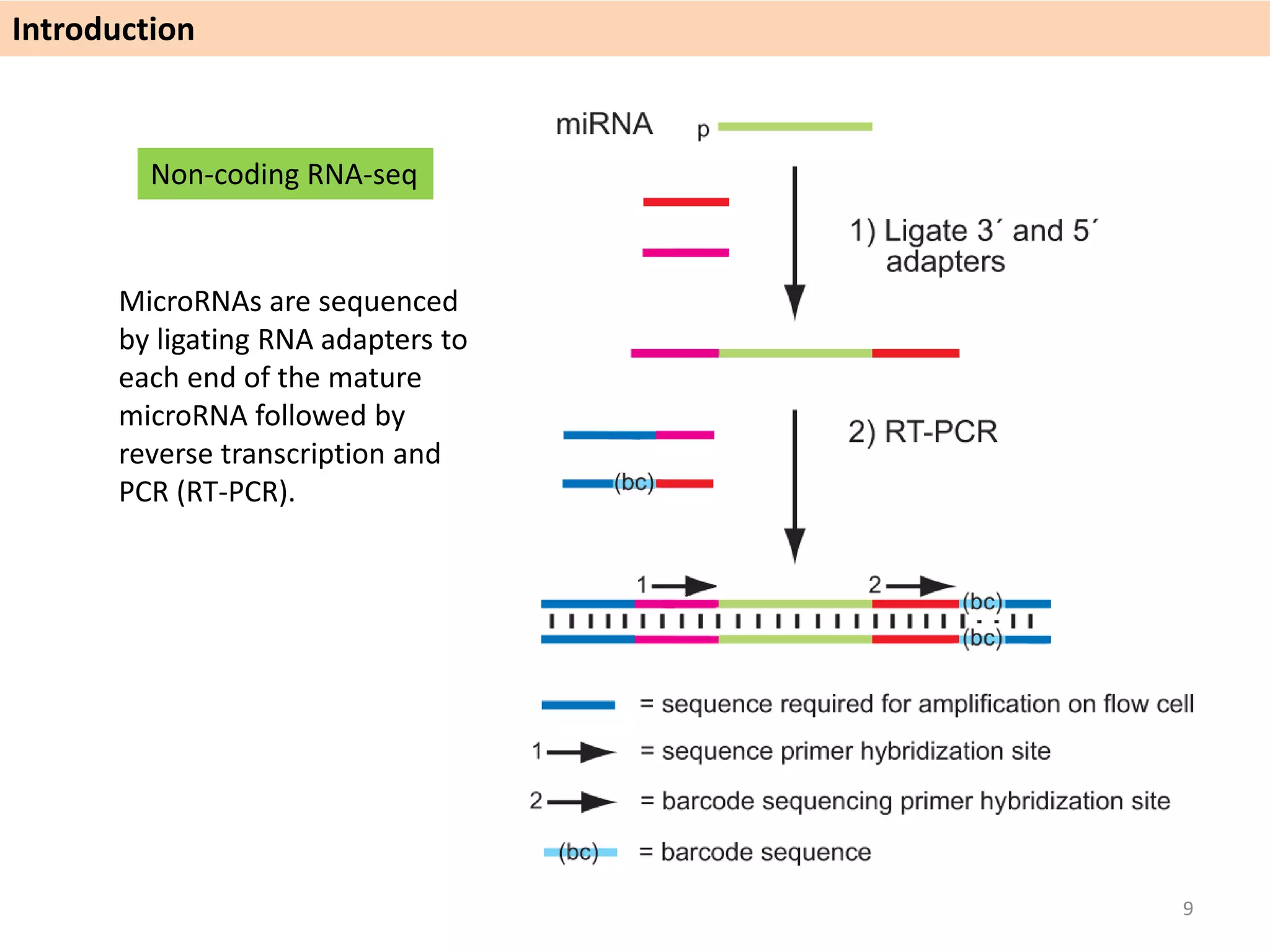
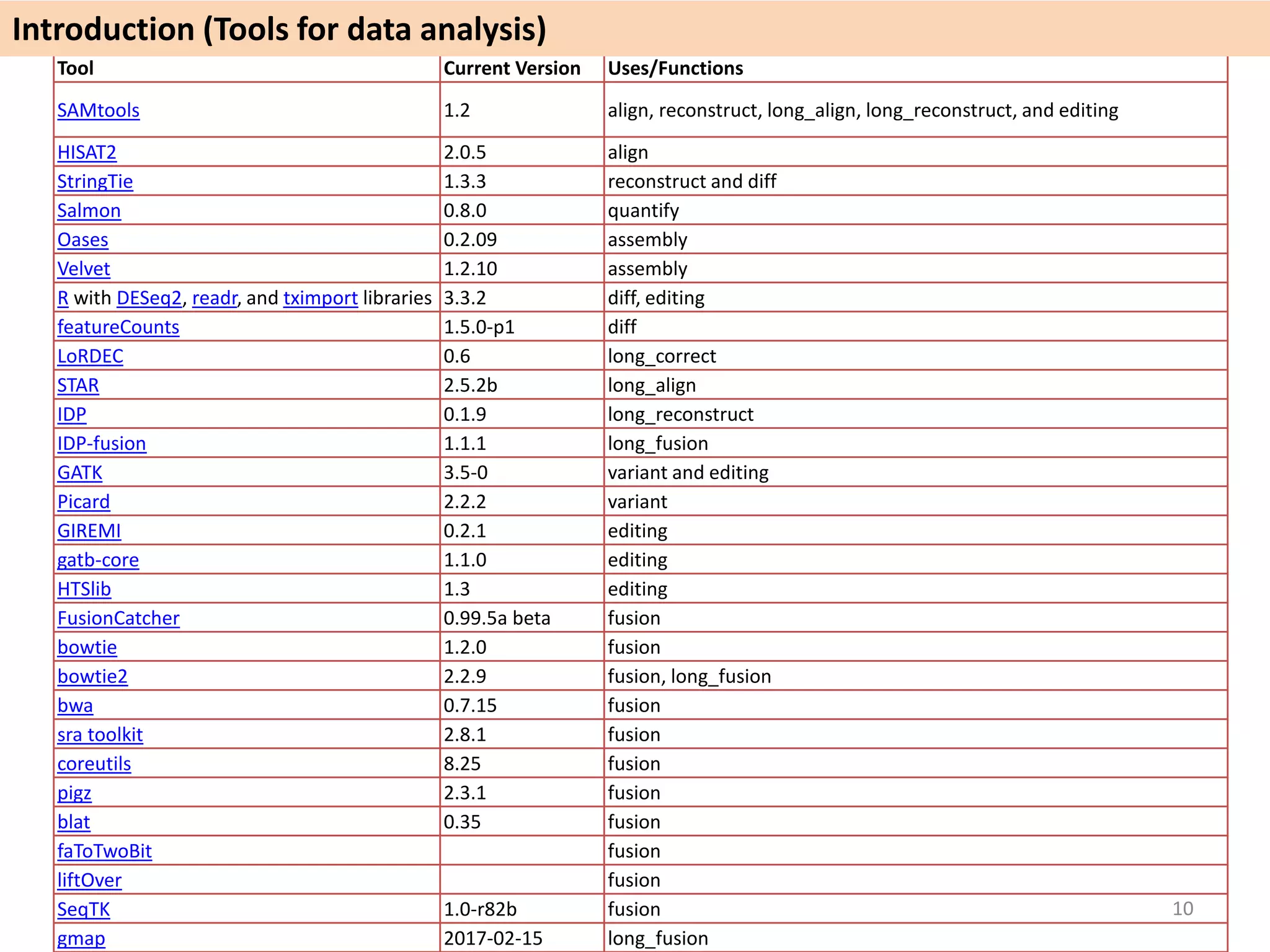
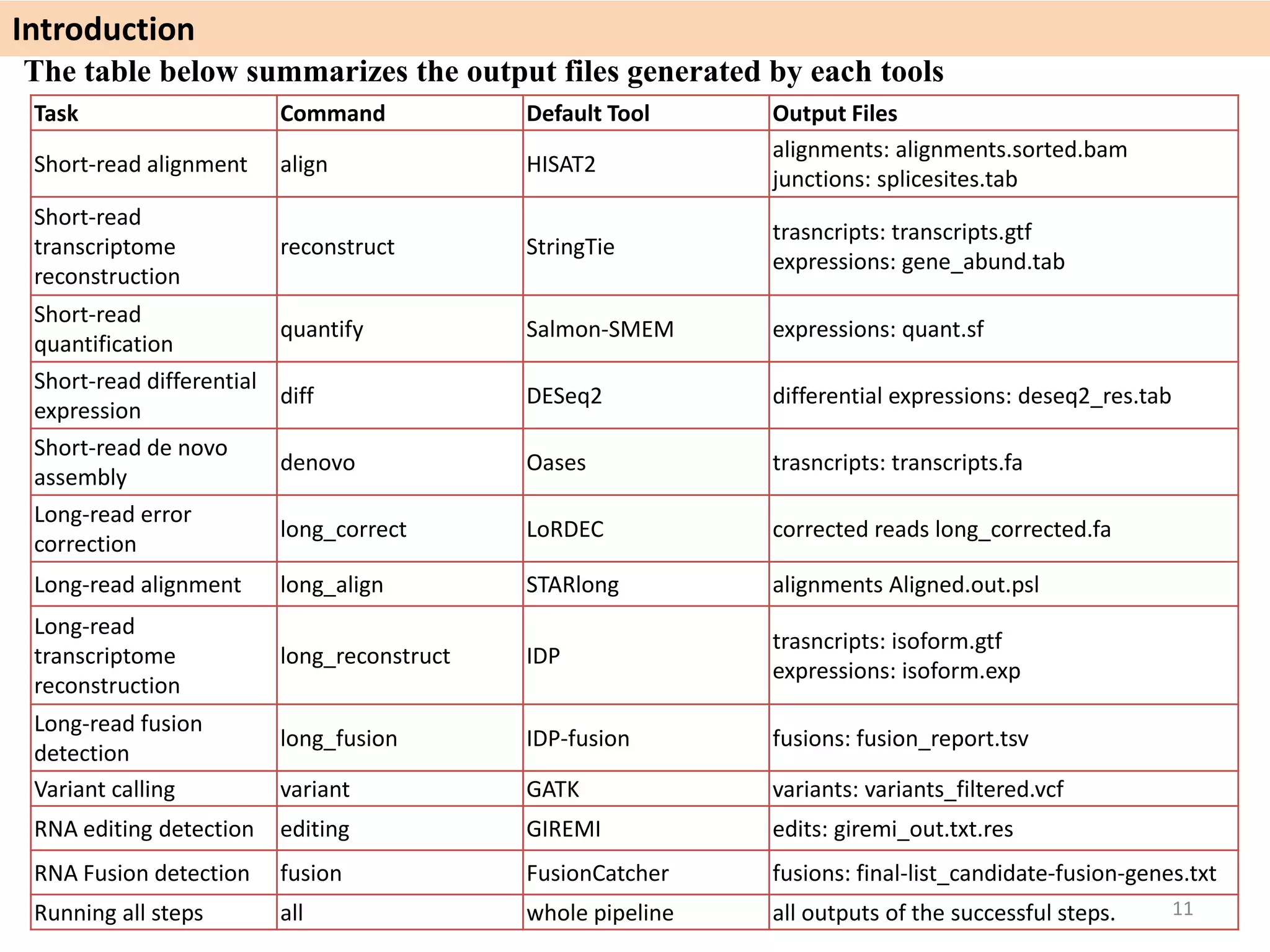


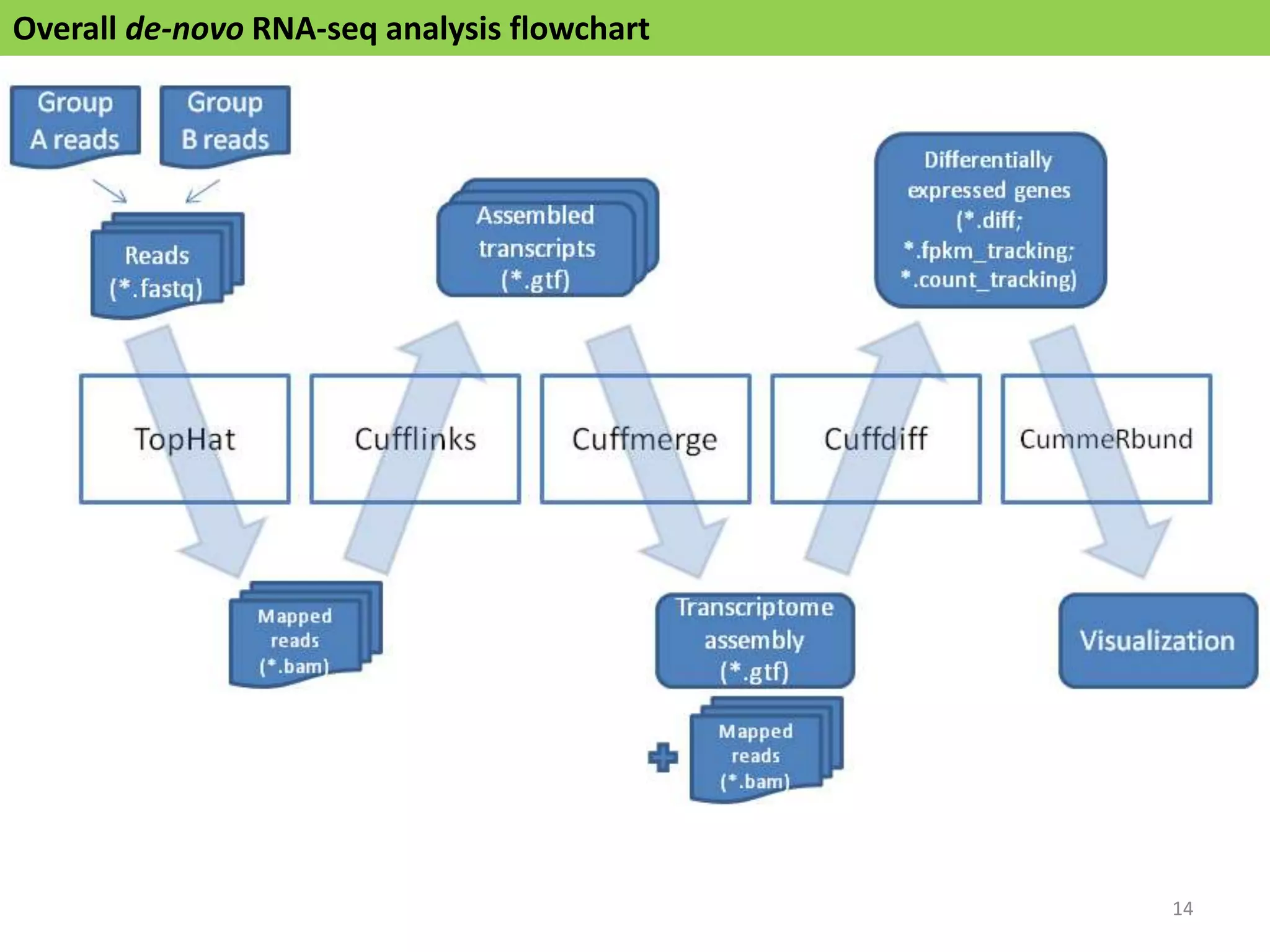
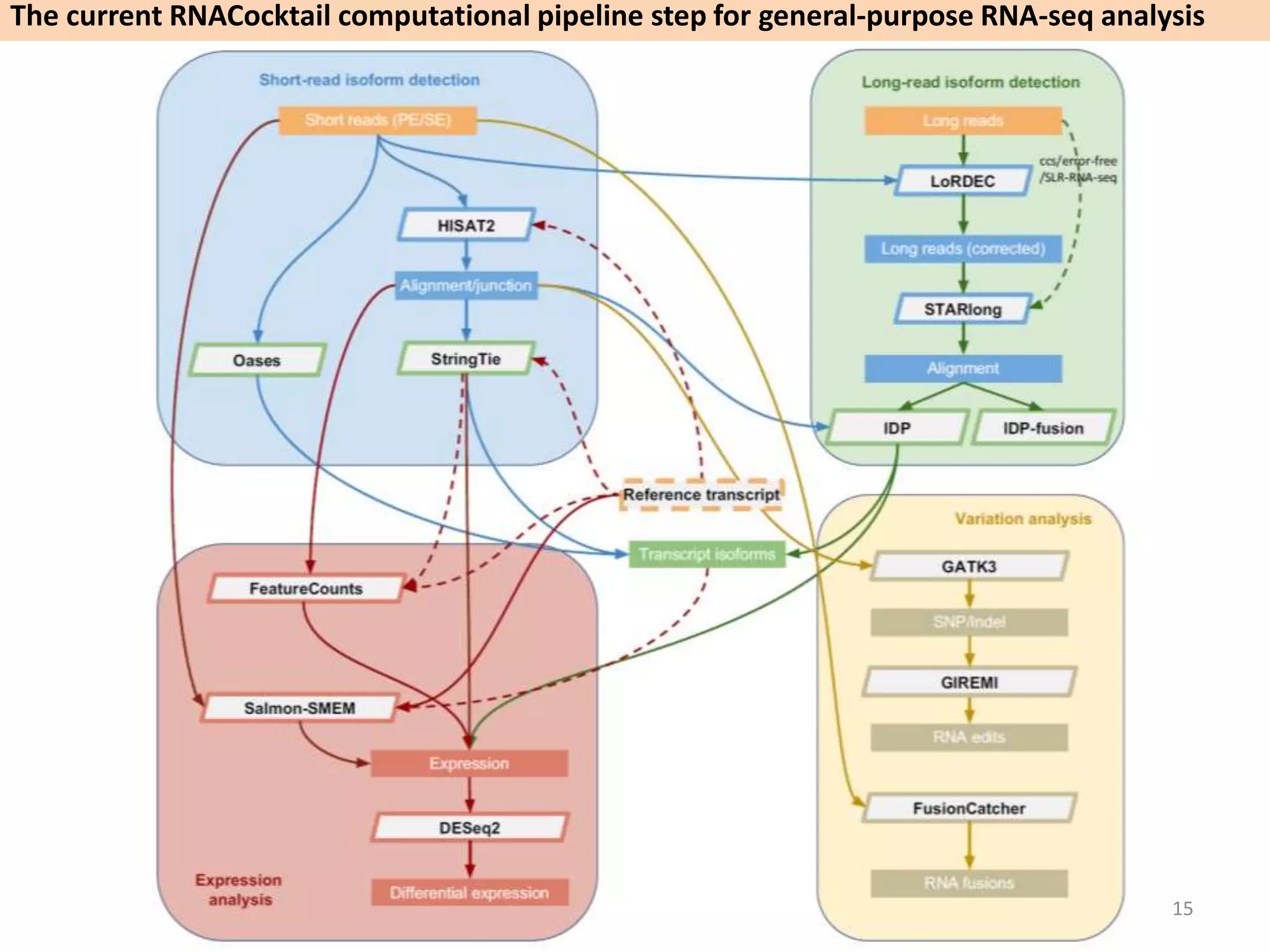

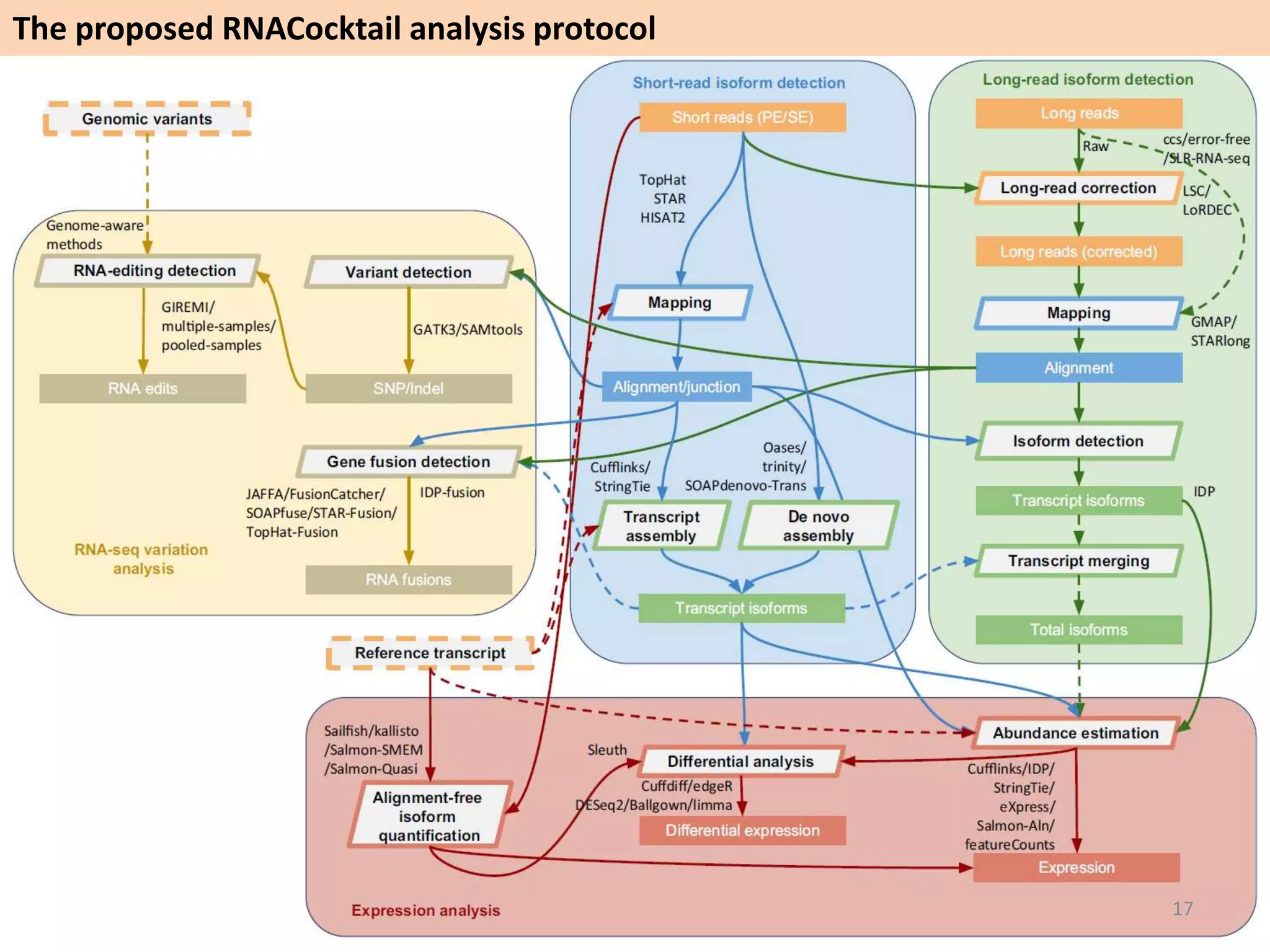
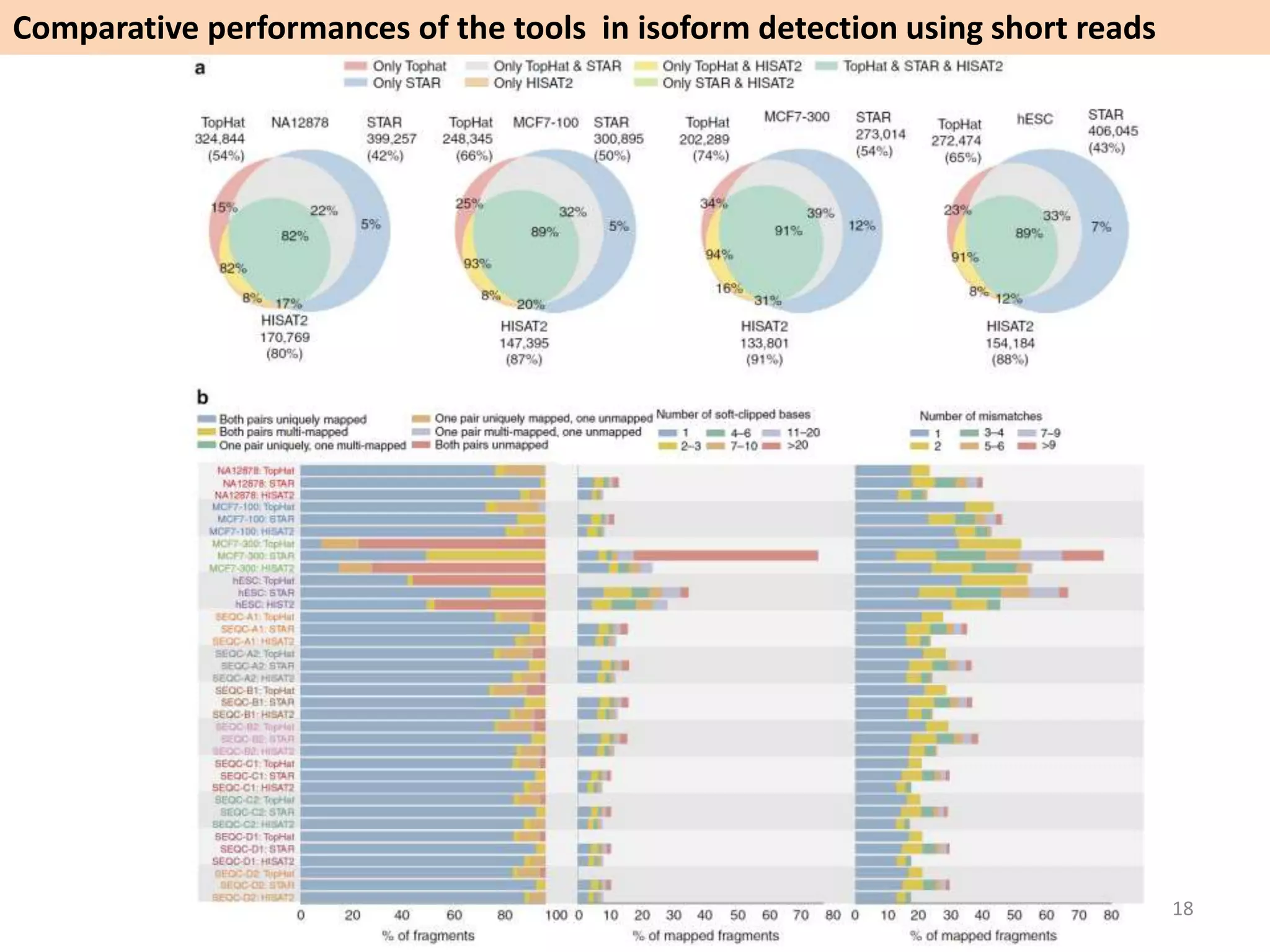

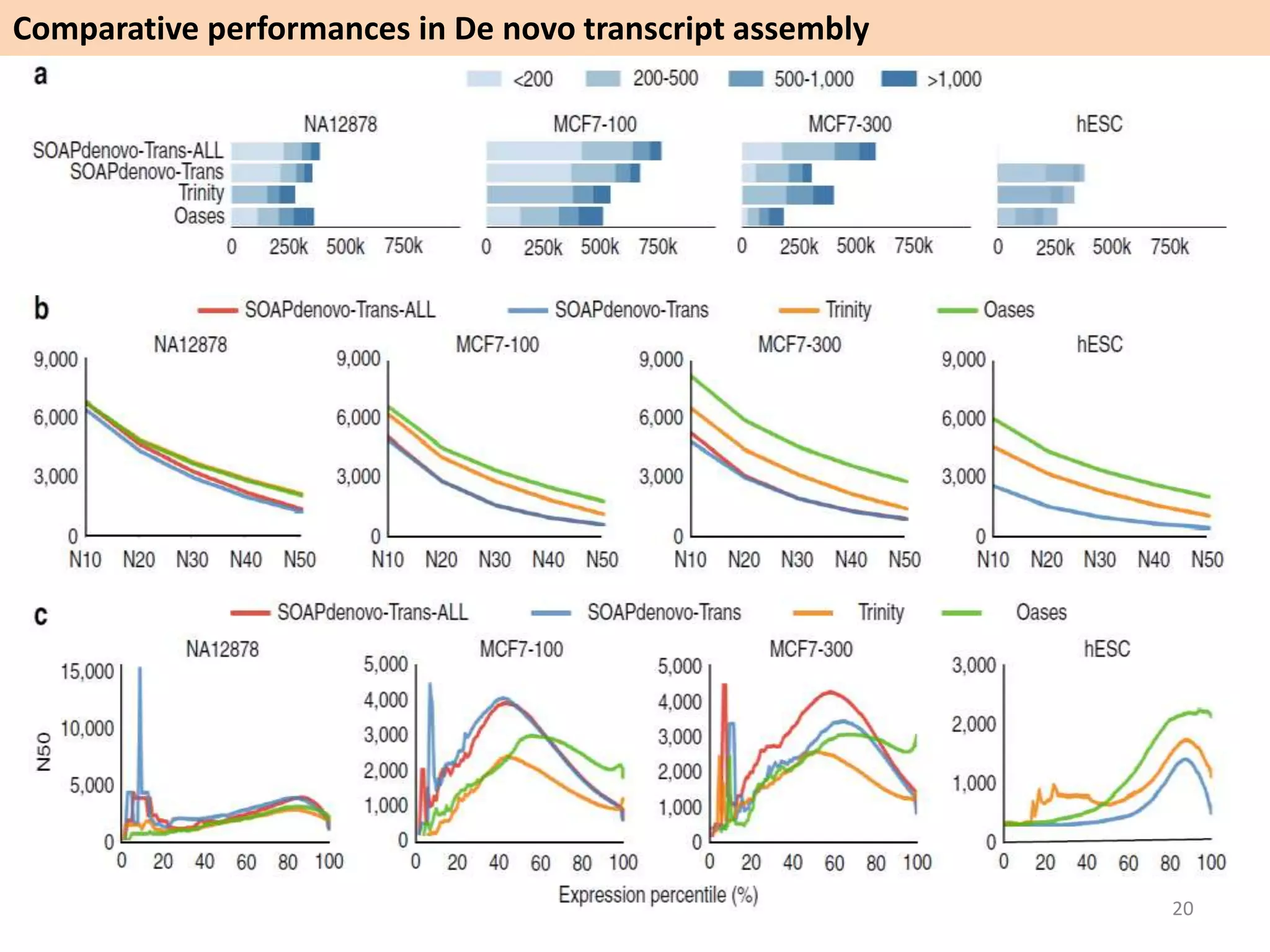
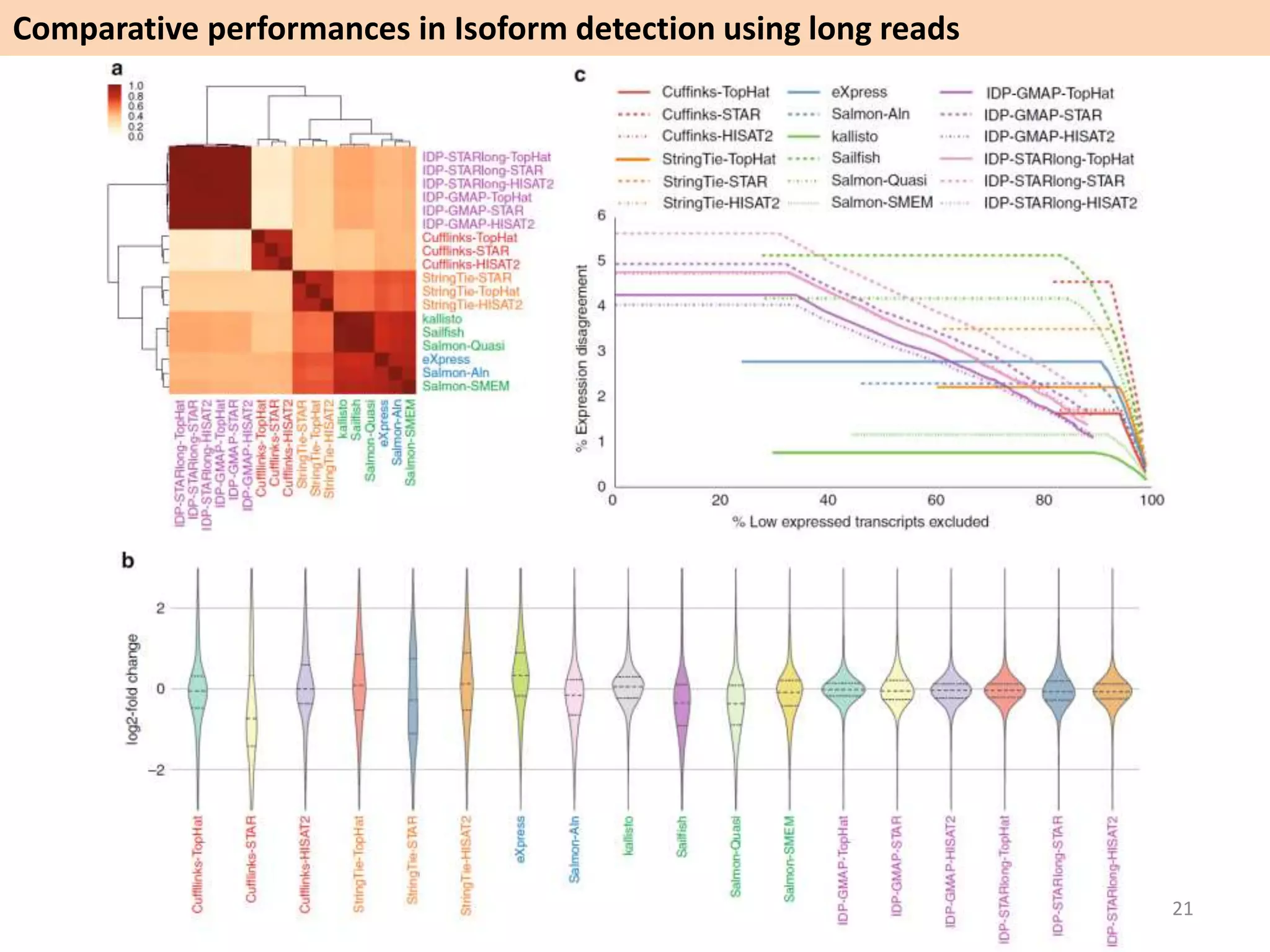
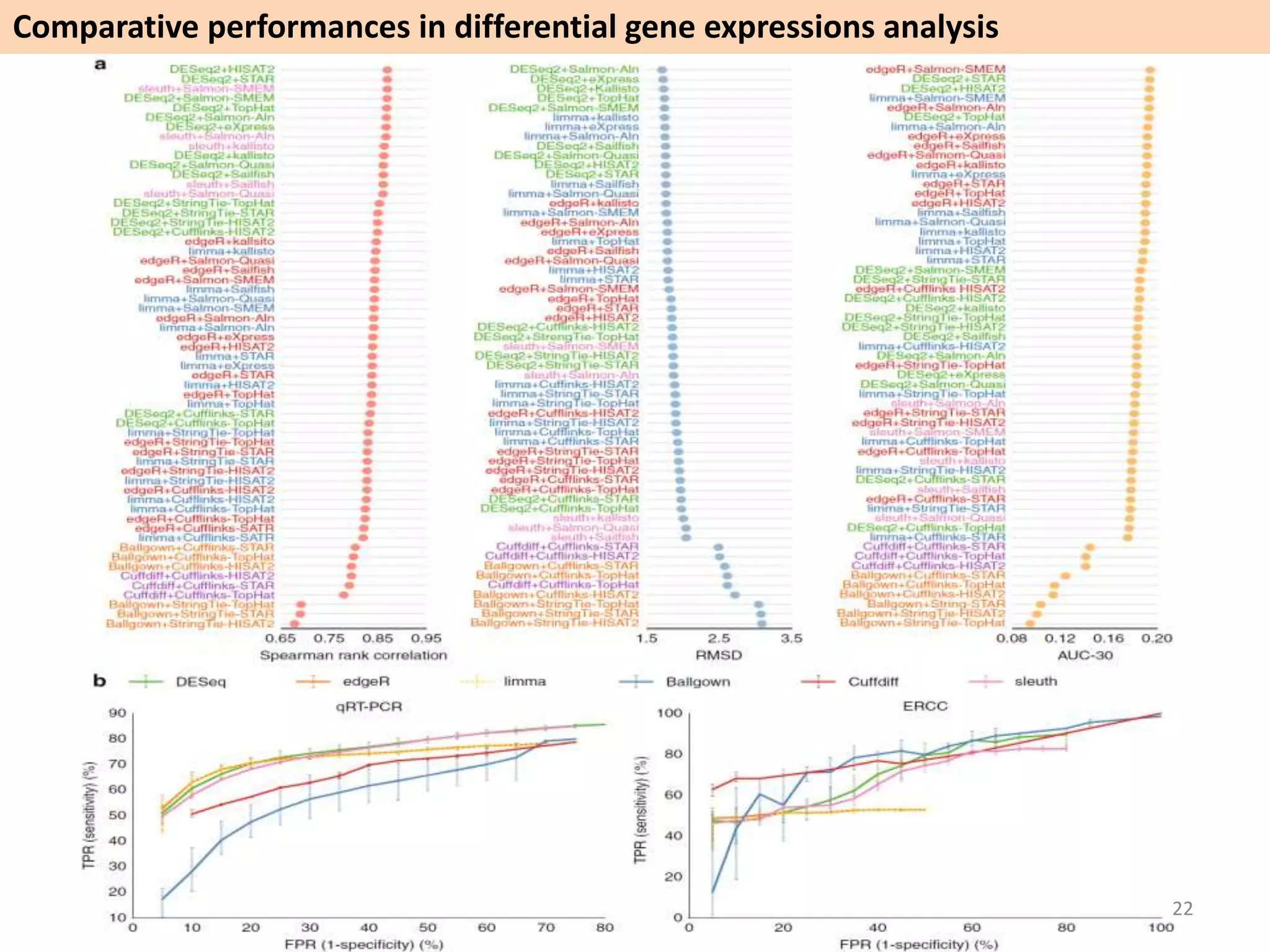
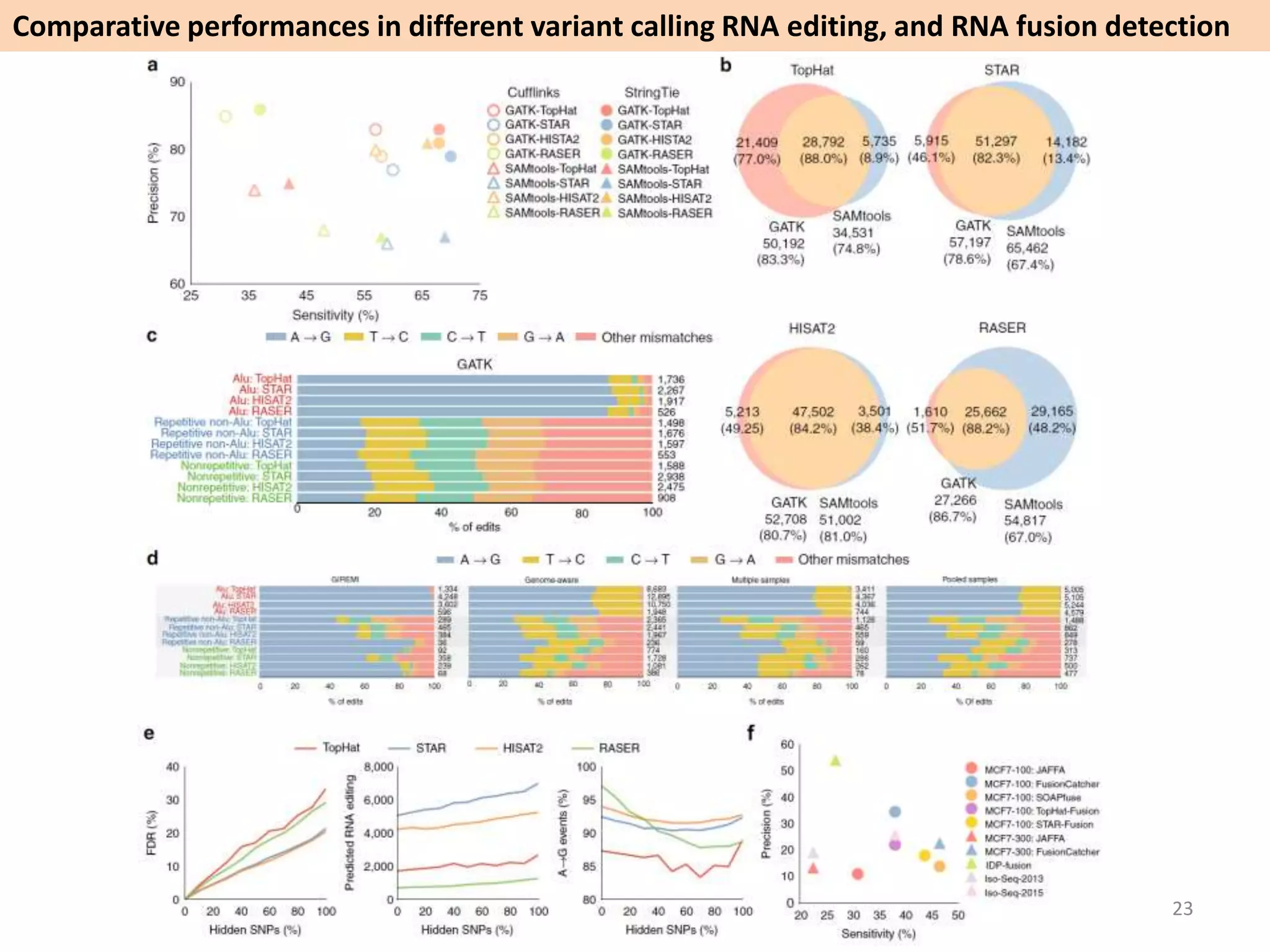



The document describes an RNA-seq analysis workflow called RNACocktail. It evaluates tools for various RNA-seq analysis tasks like alignment, assembly, quantification, and more. The study finds that performance can vary significantly depending on the specific tool and data set. It then proposes a comprehensive RNACocktail protocol and computational pipeline that achieves high accuracy across different sample types and analysis goals. Validation on multiple samples shows this broad analysis approach can help researchers extract more biologically relevant insights from transcriptomic data.