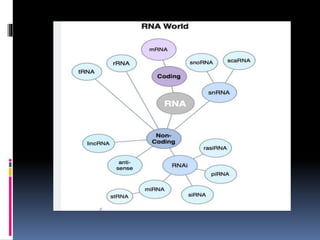
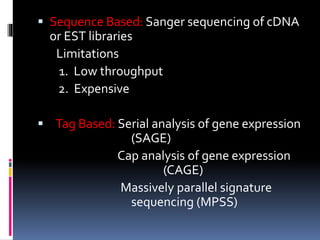
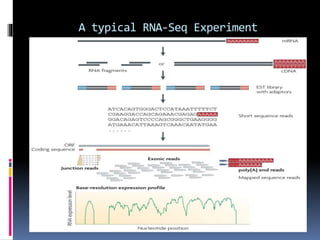
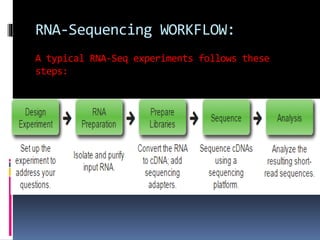
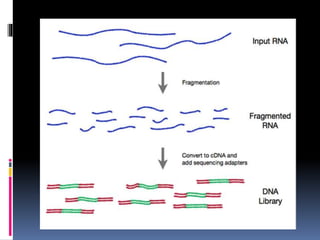
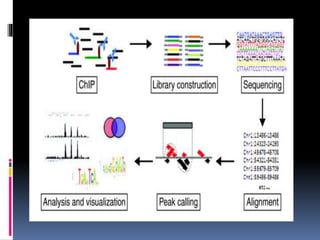
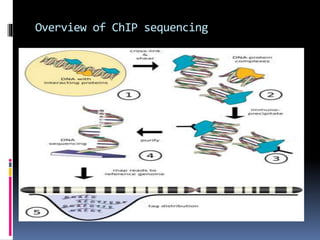
This document provides an overview of RNA sequencing (RNA-Seq) and chromatin immunoprecipitation sequencing (ChIP-Seq). It describes that RNA-Seq is used to profile transcriptomes and determine gene expression levels, while ChIP-Seq identifies the binding sites of DNA-associated proteins. The key steps of RNA-Seq are RNA preparation, library preparation, sequencing, and analysis to map reads, detect isoforms and expression levels. ChIP-Seq combines chromatin immunoprecipitation with sequencing to precisely map global binding sites of proteins of interest to understand gene regulation. Both techniques provide high-quality, genome-wide data with low input requirements compared to previous methods.