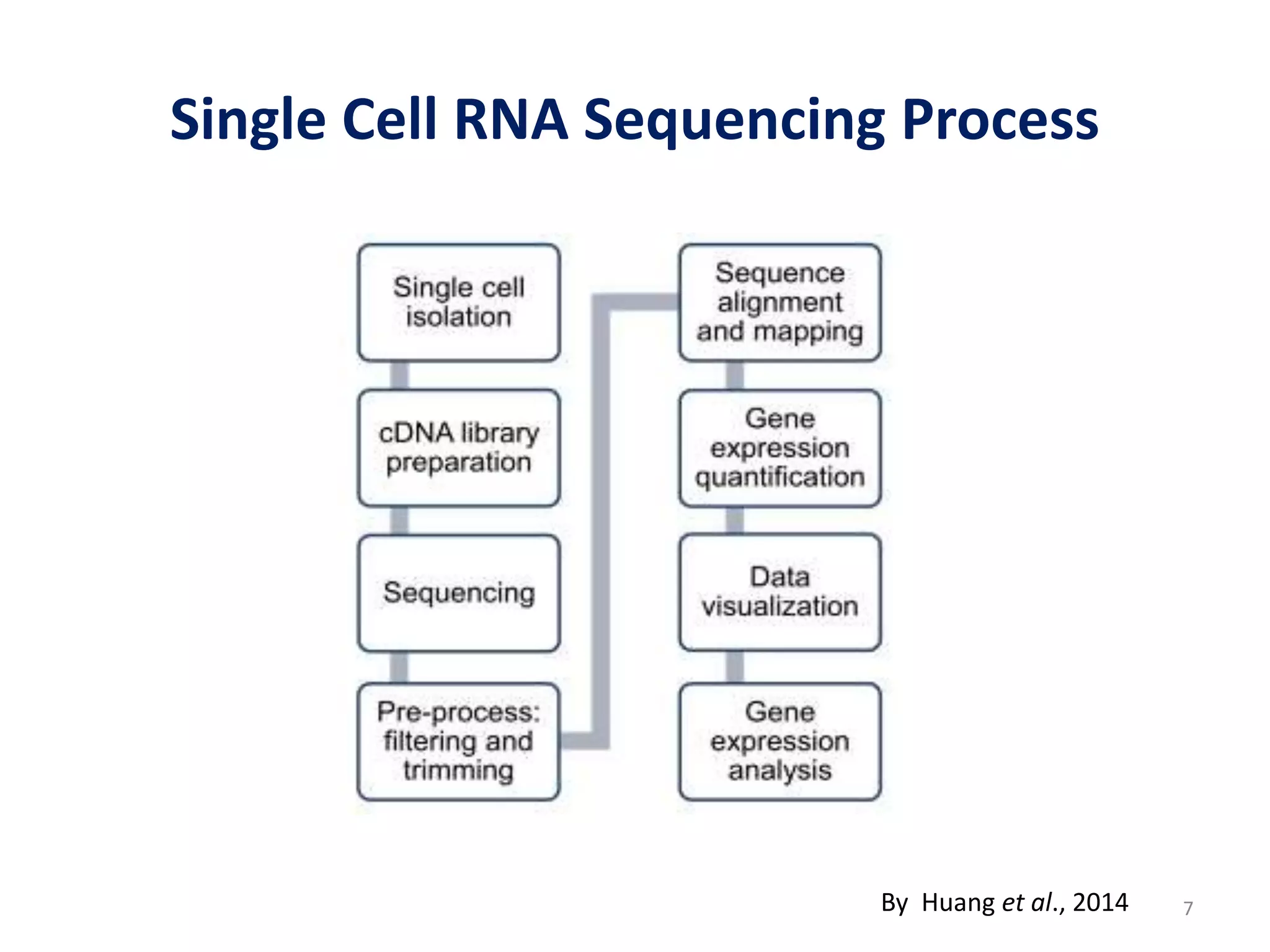
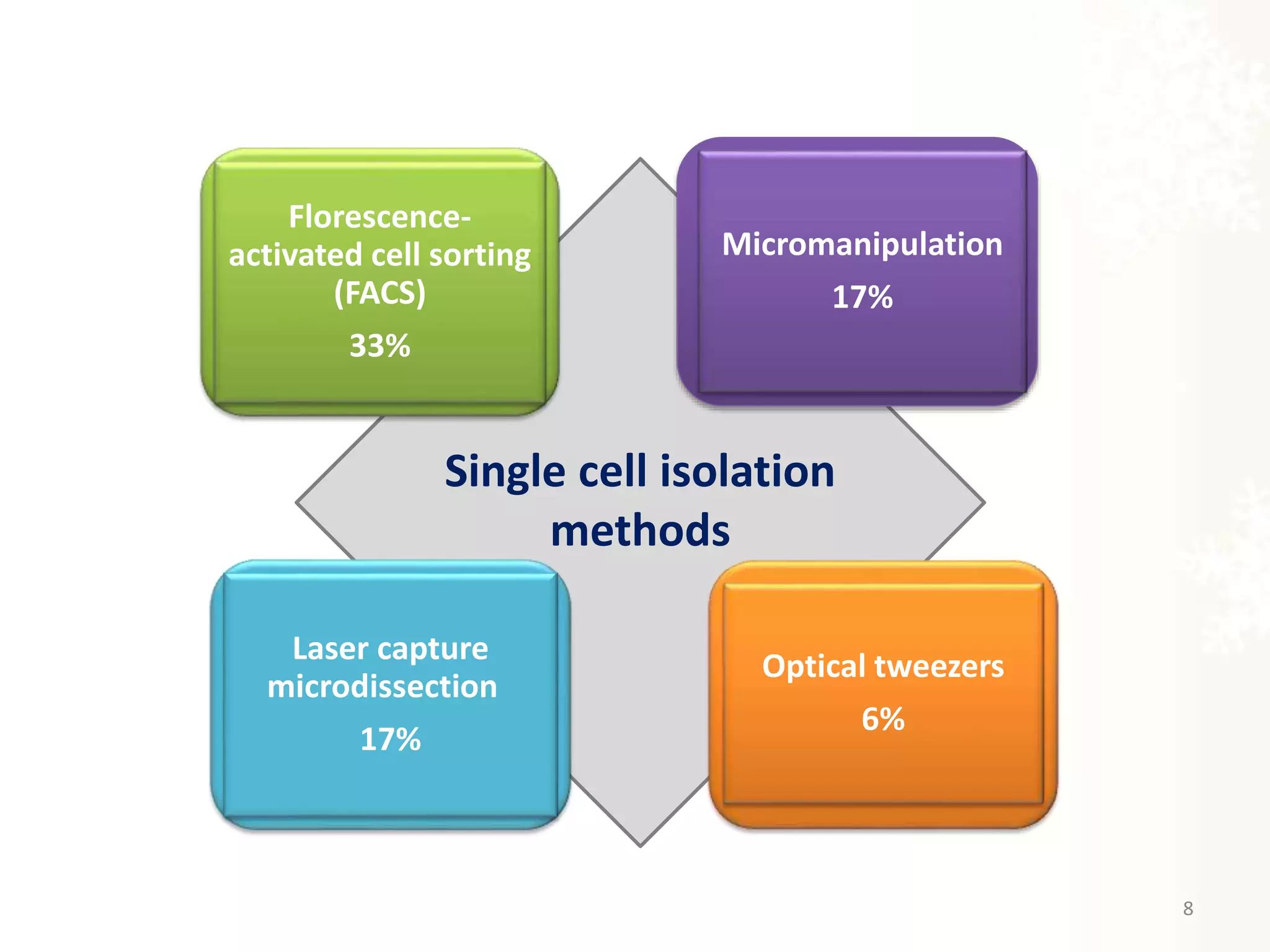
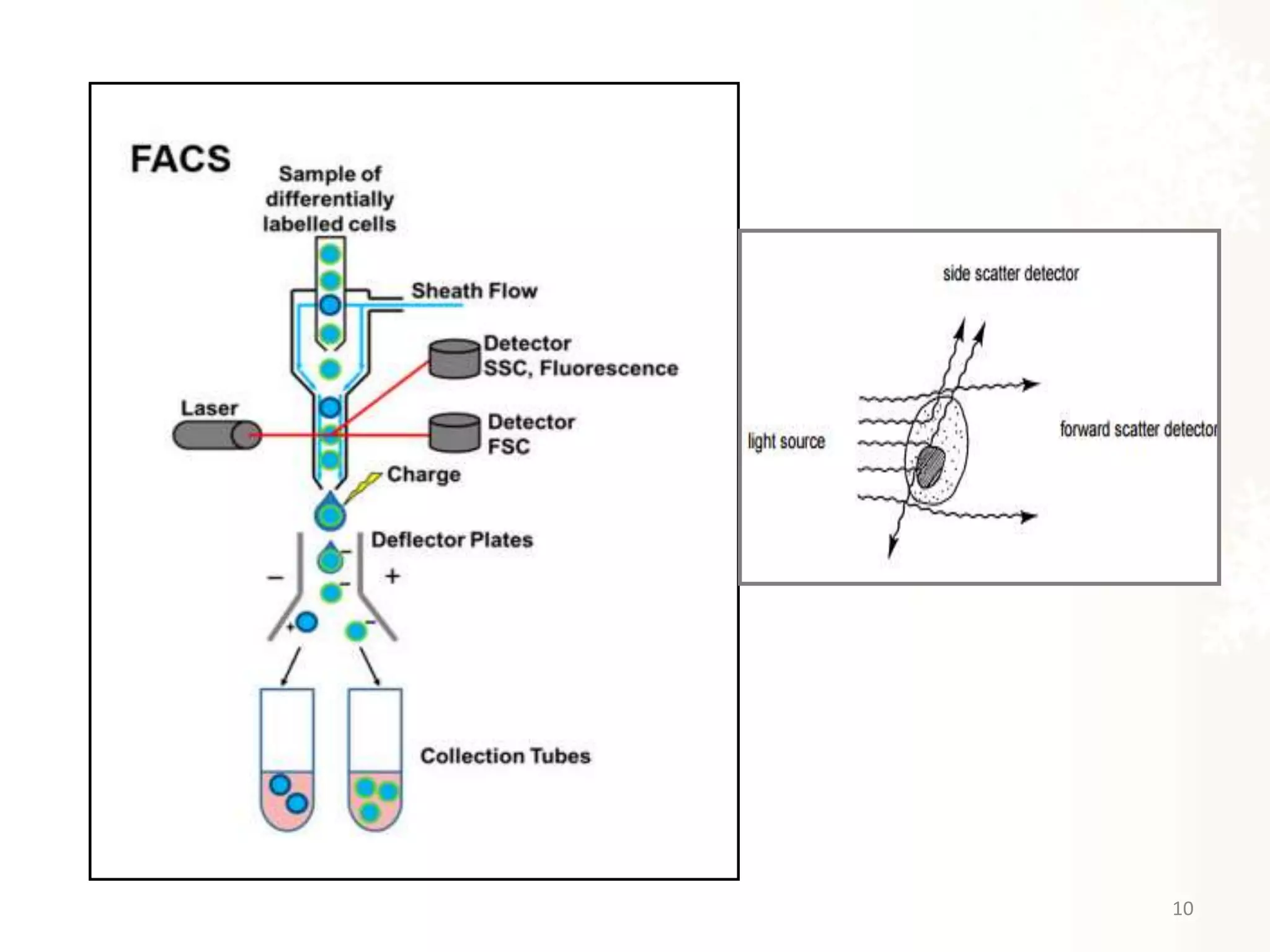
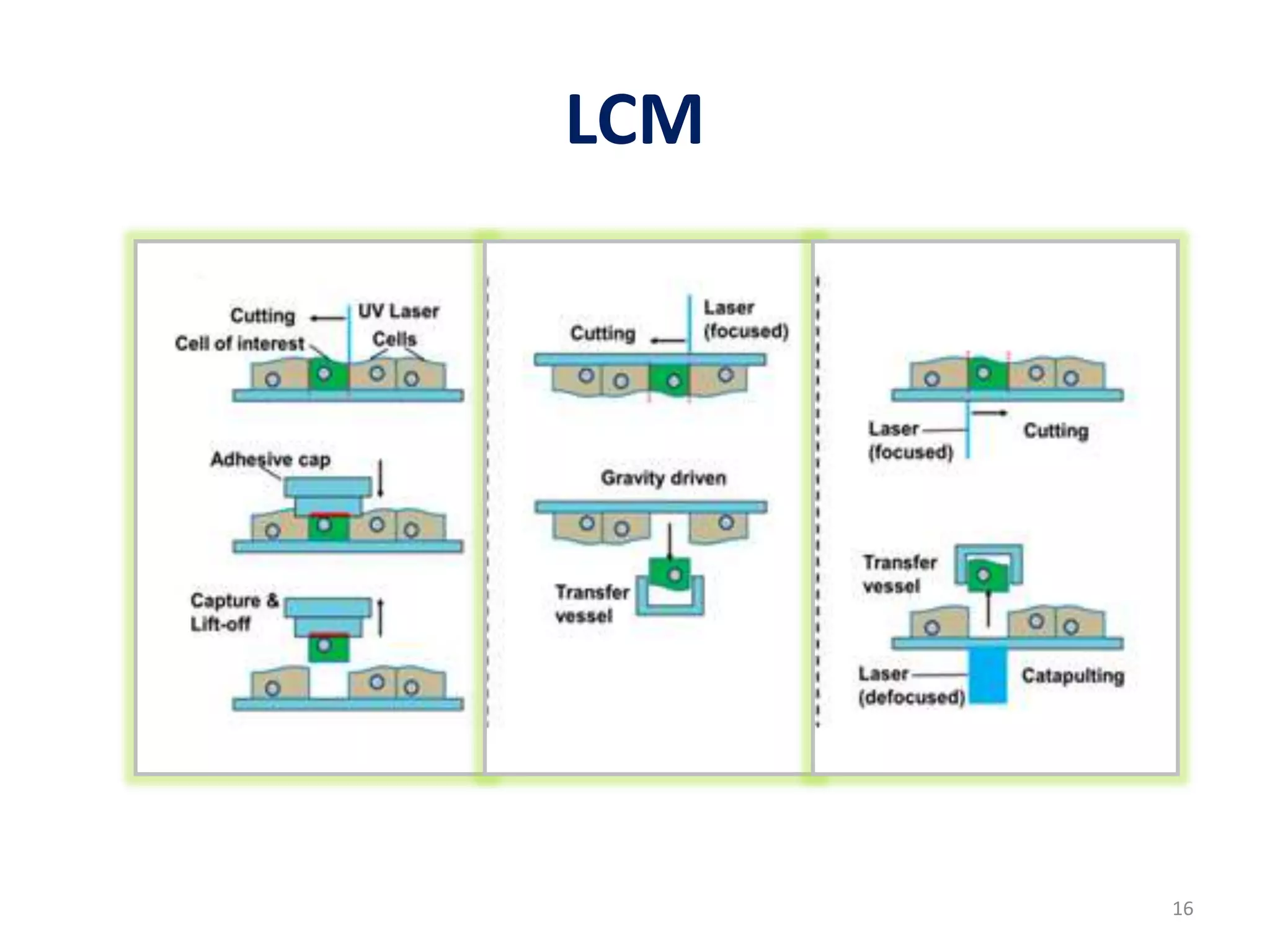
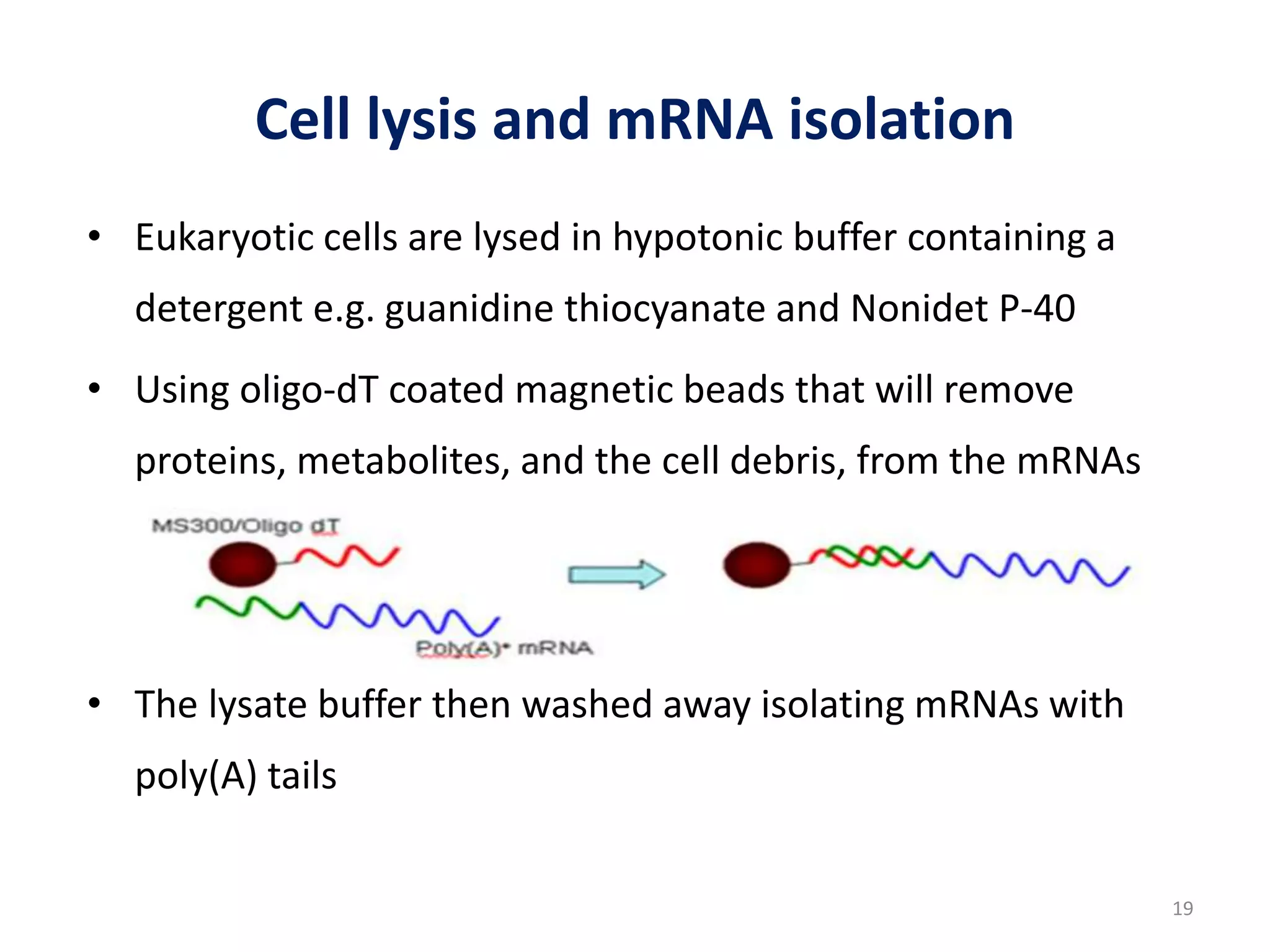
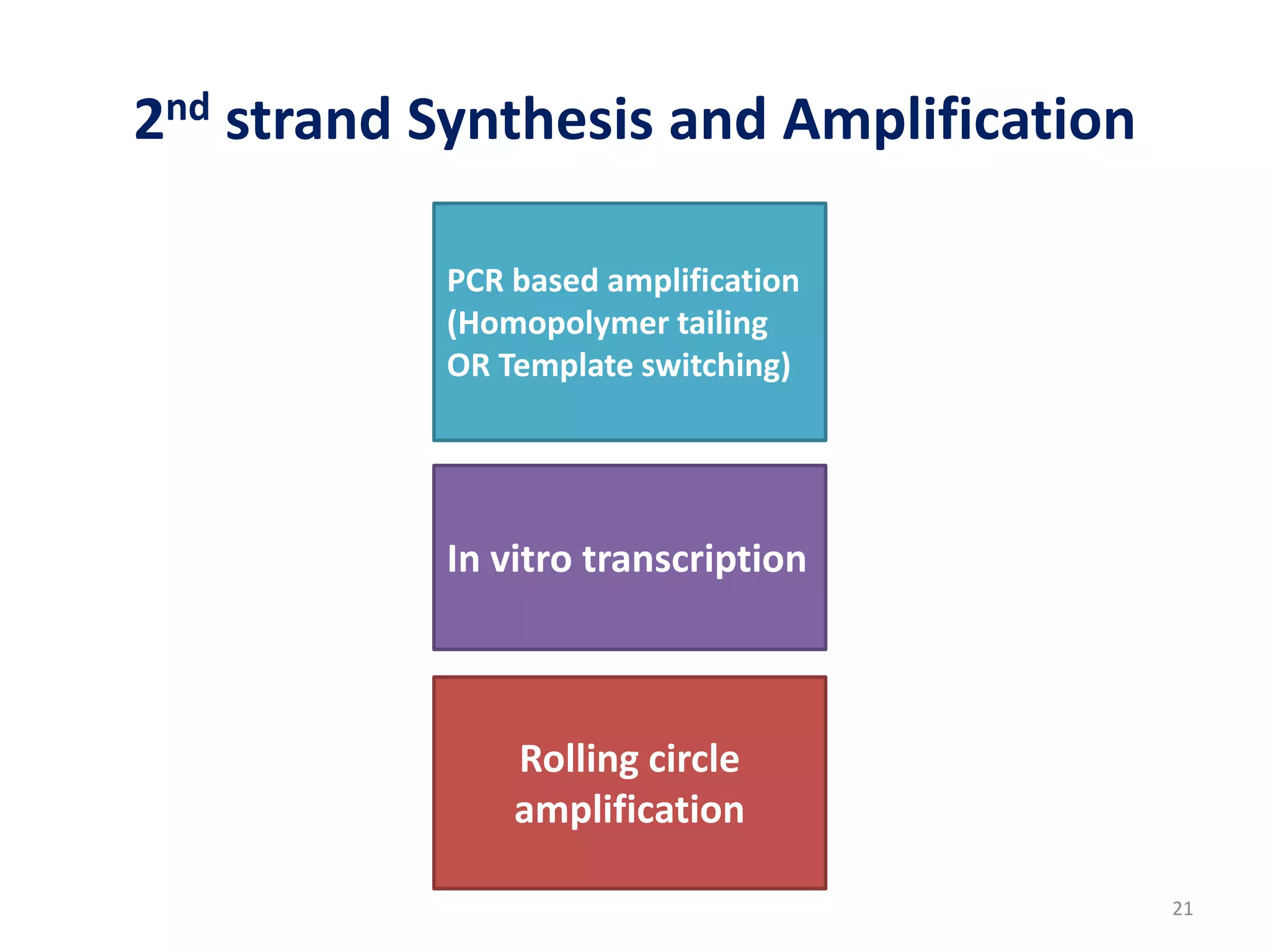
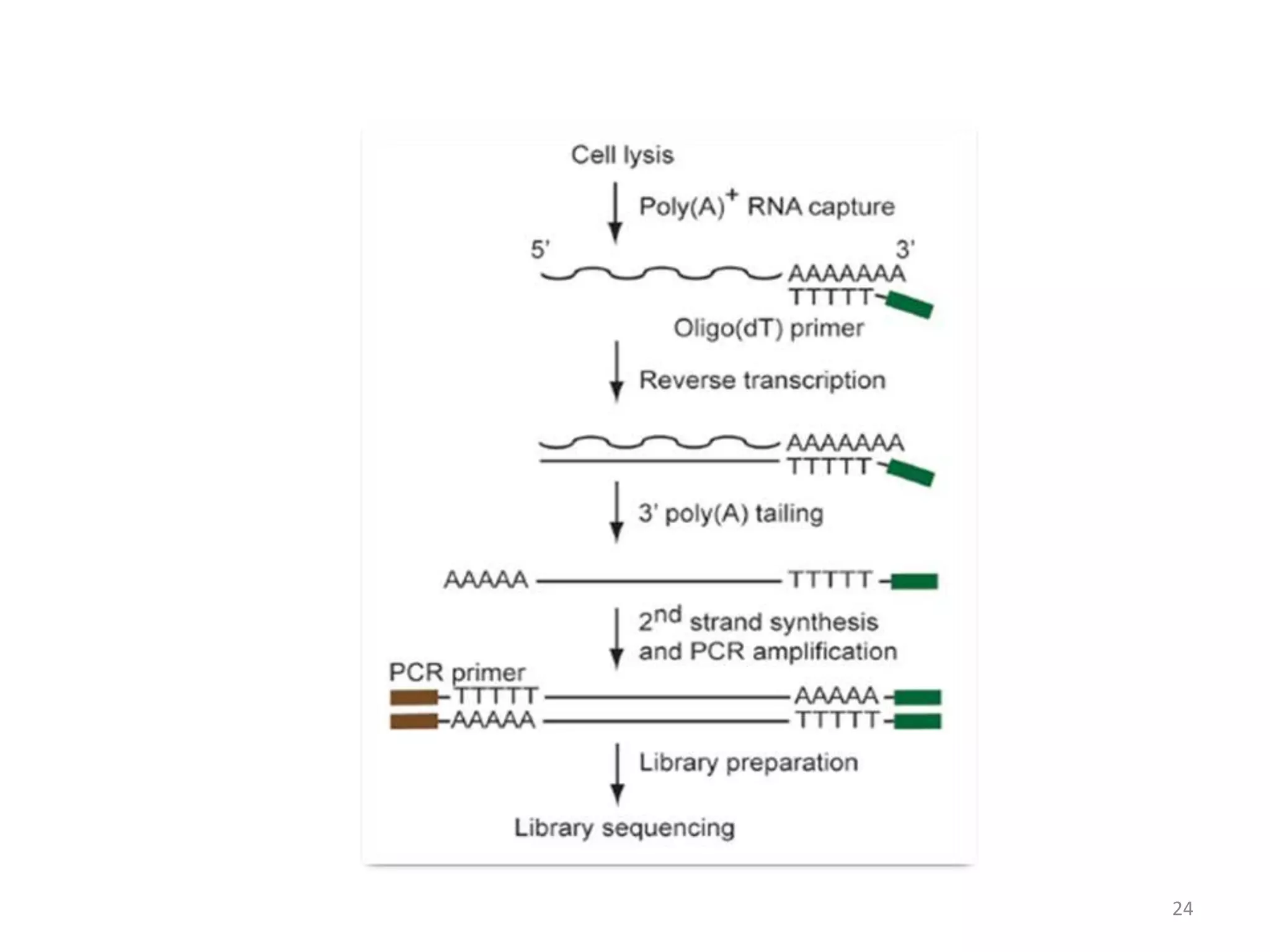
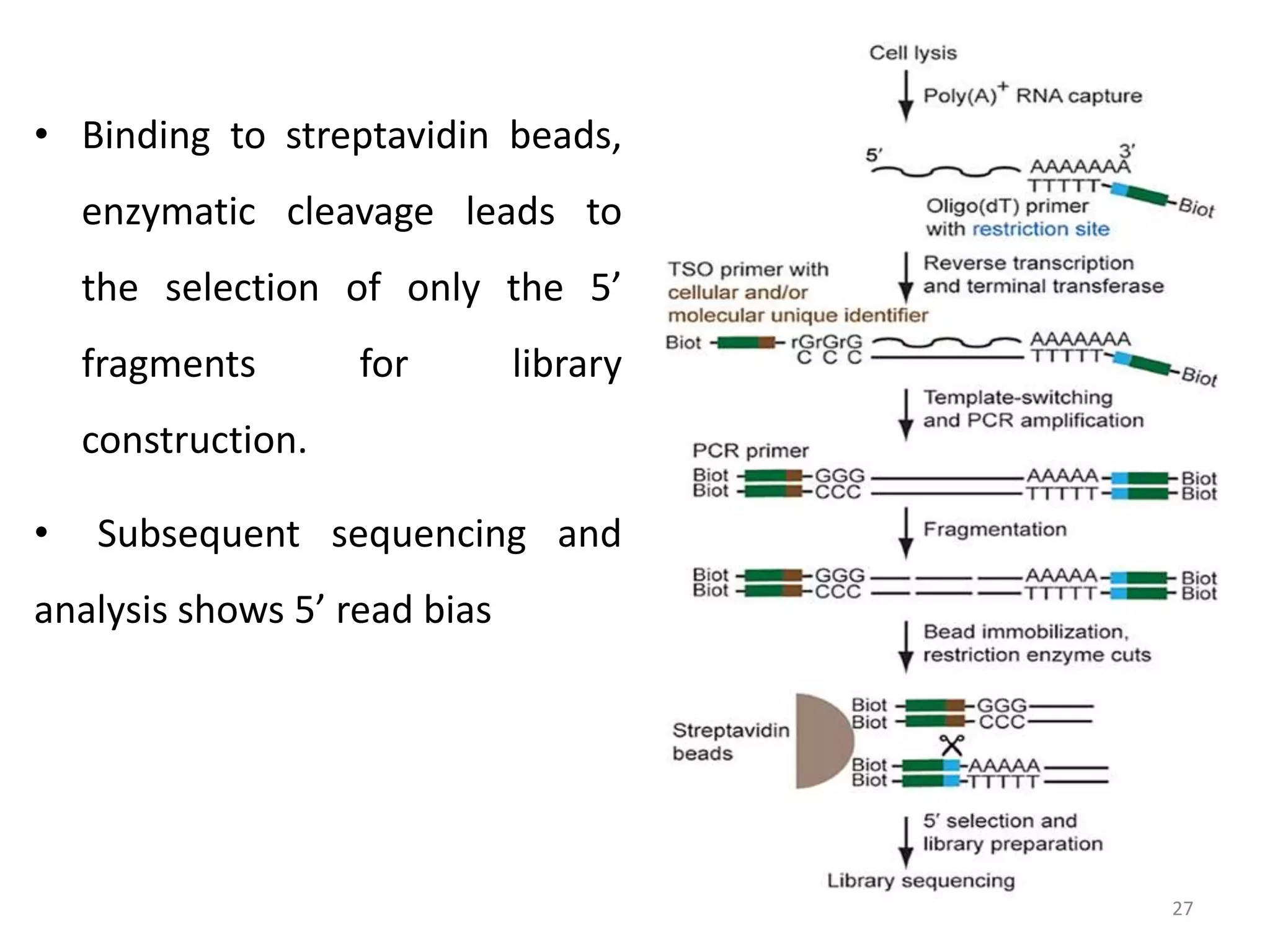
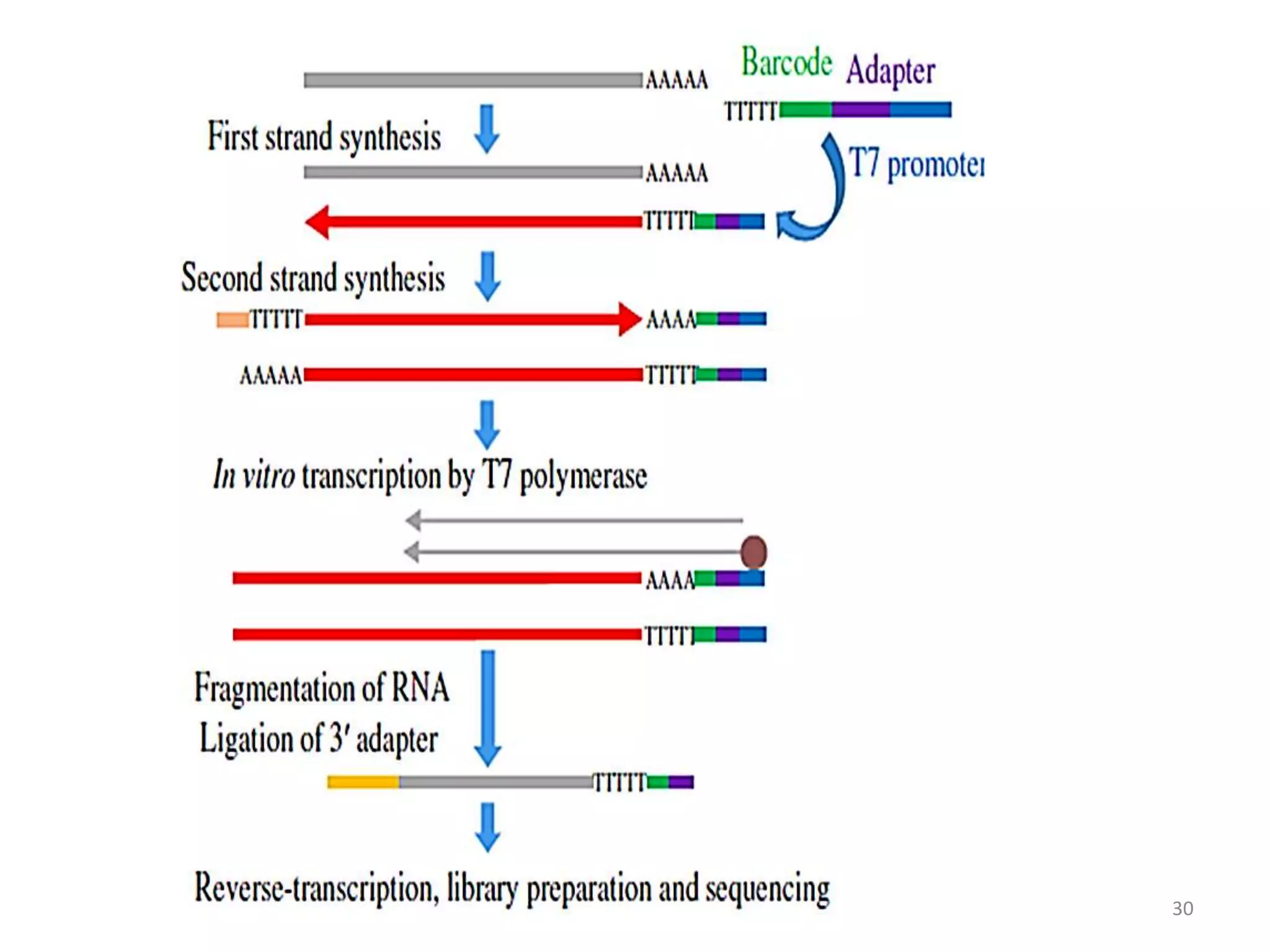
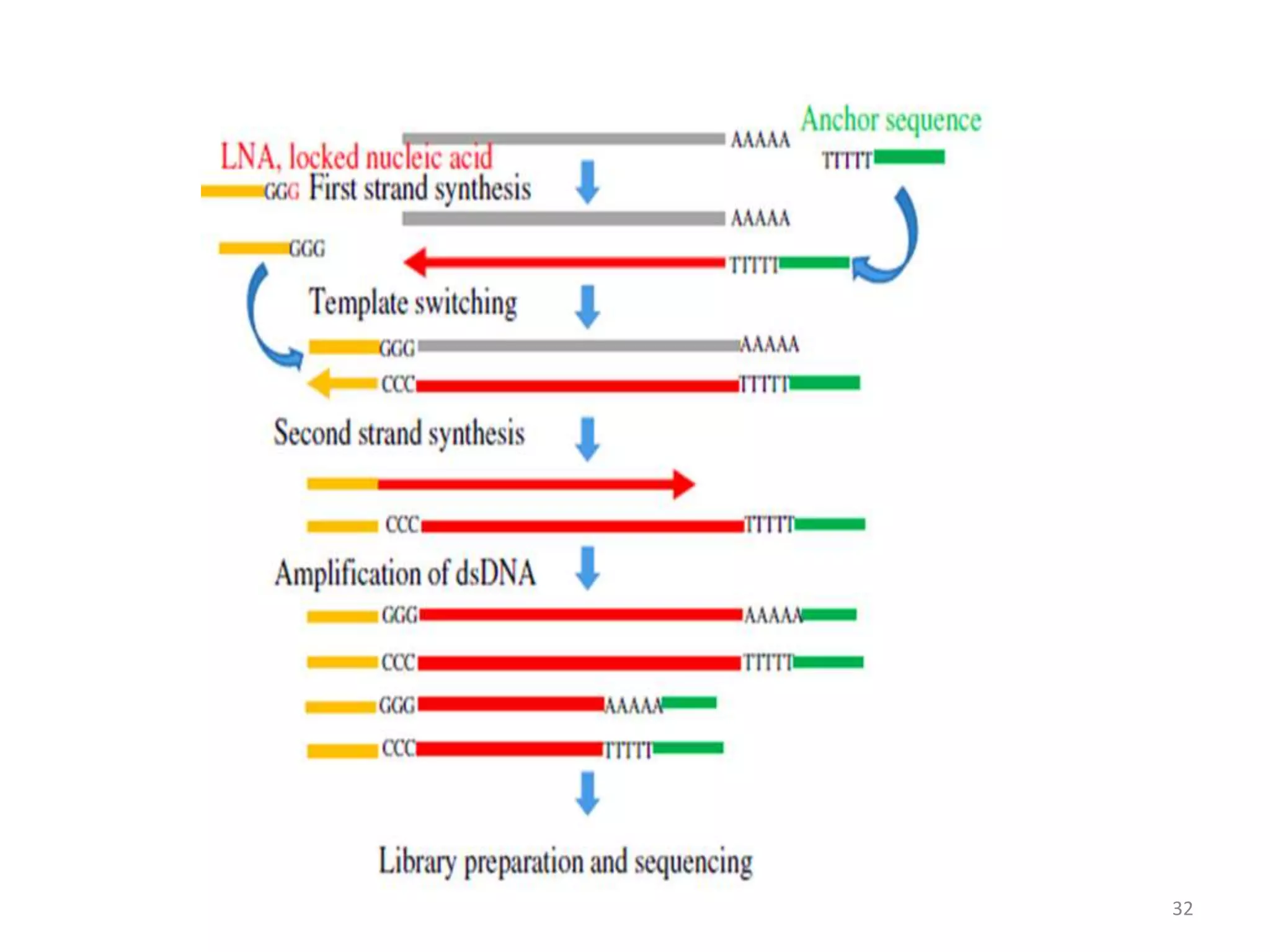
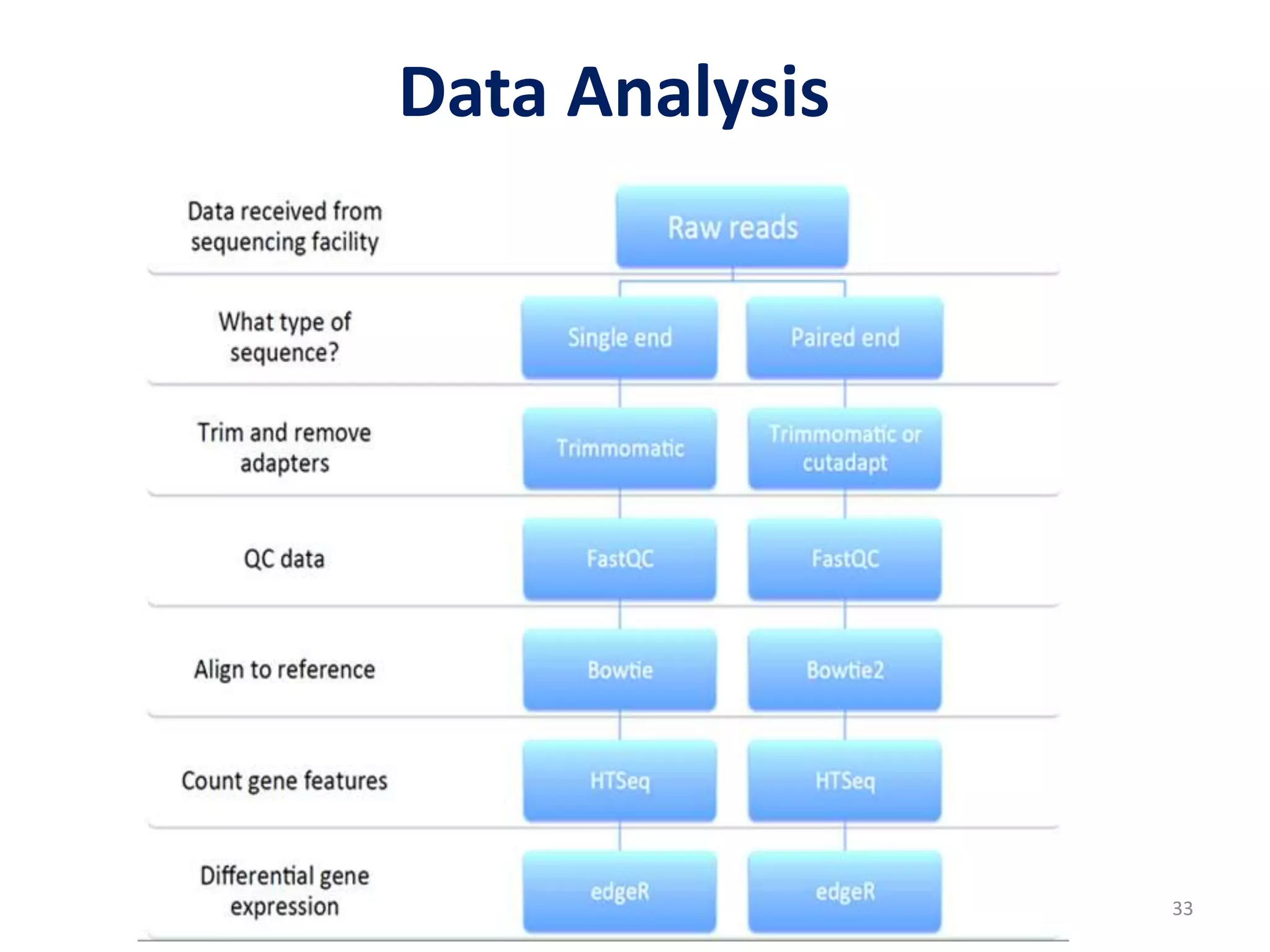
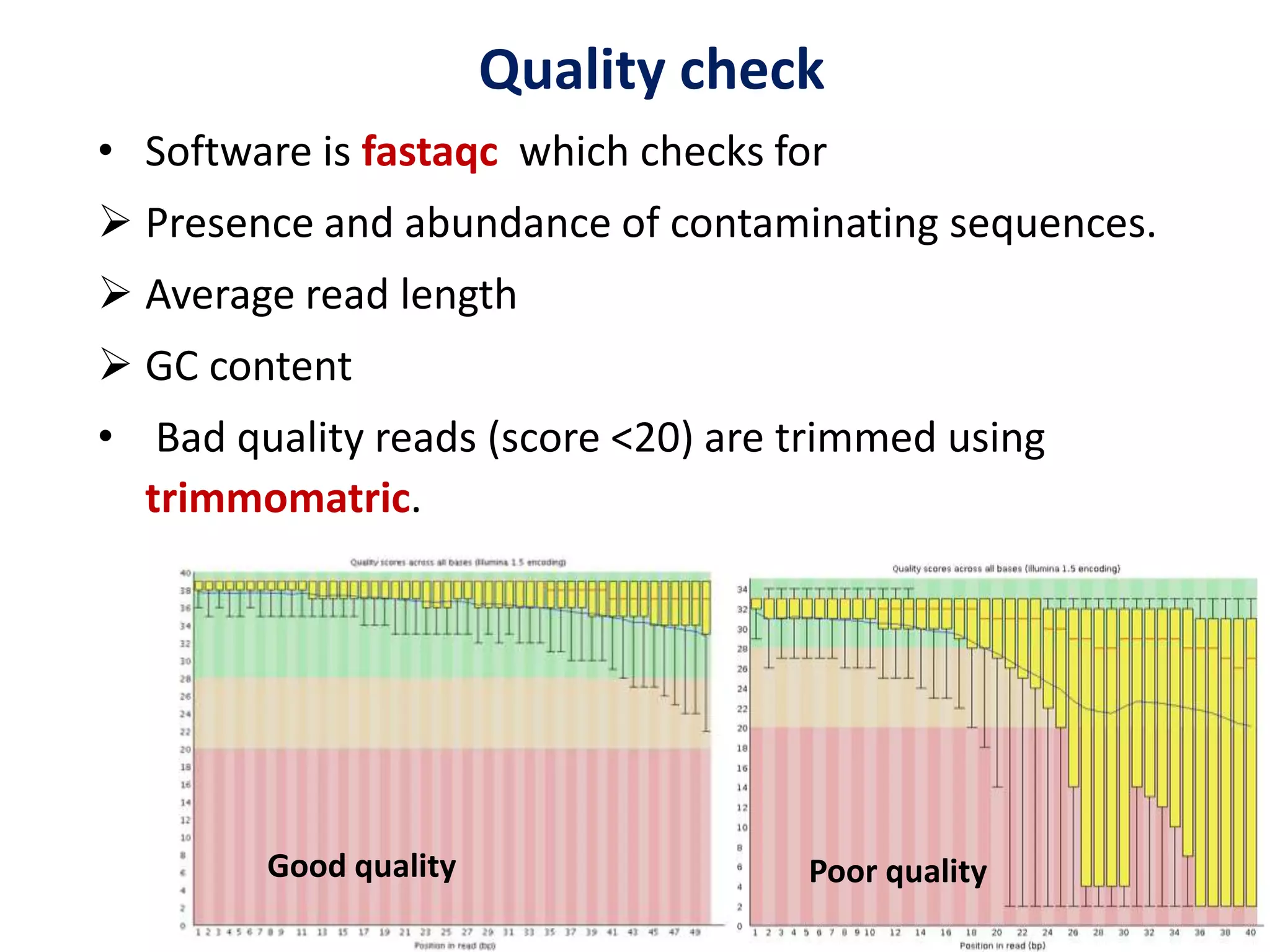
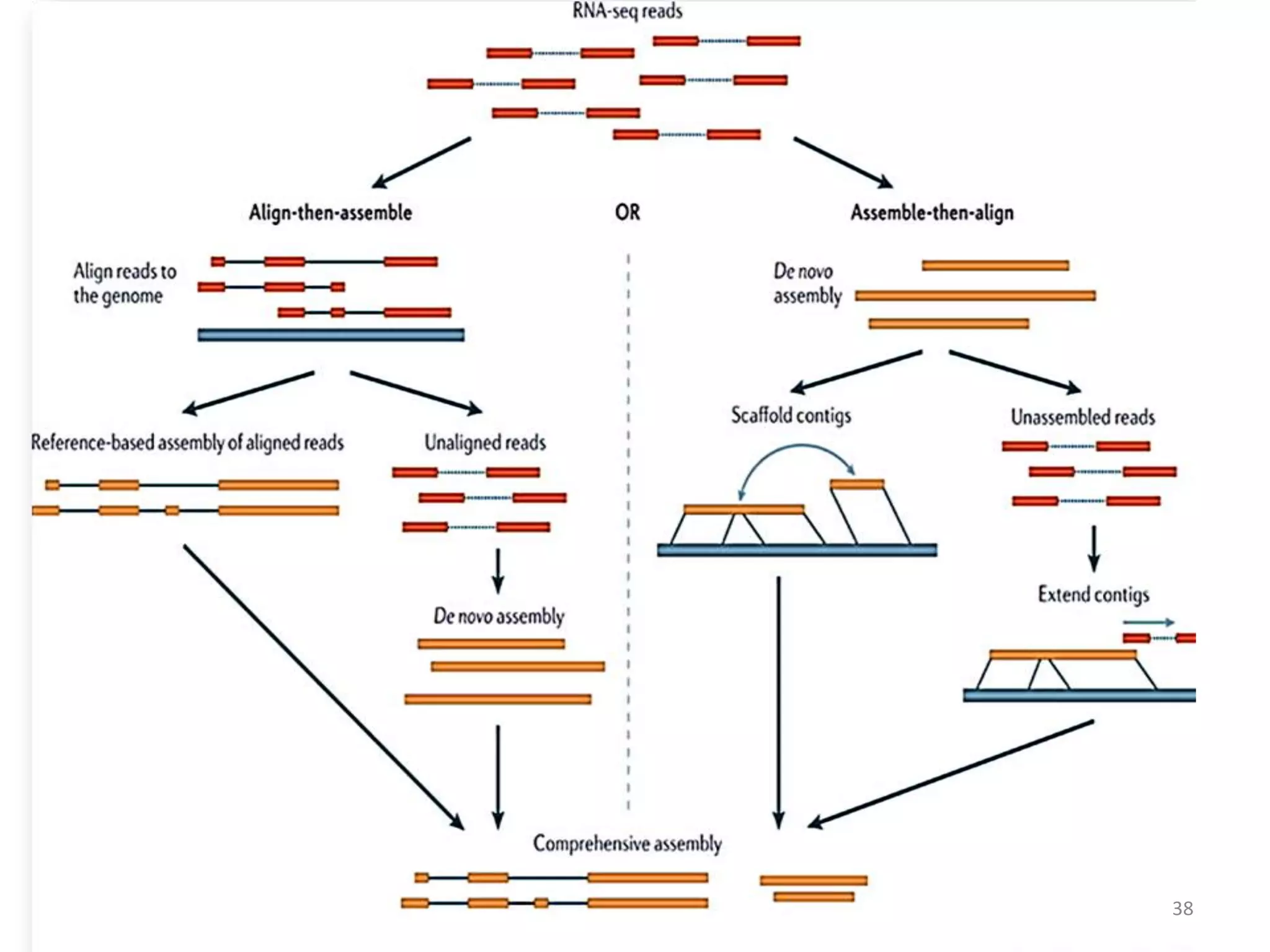
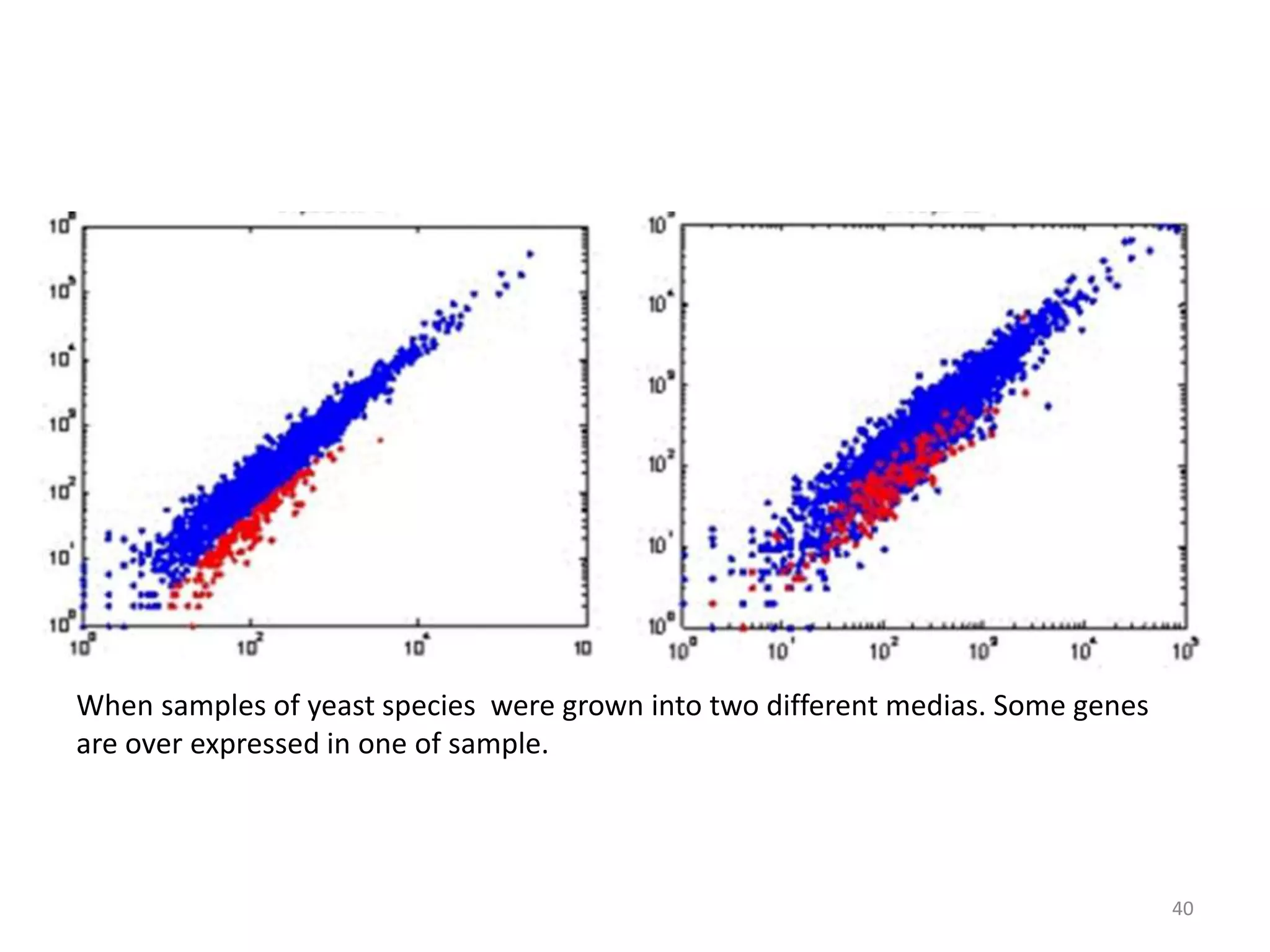
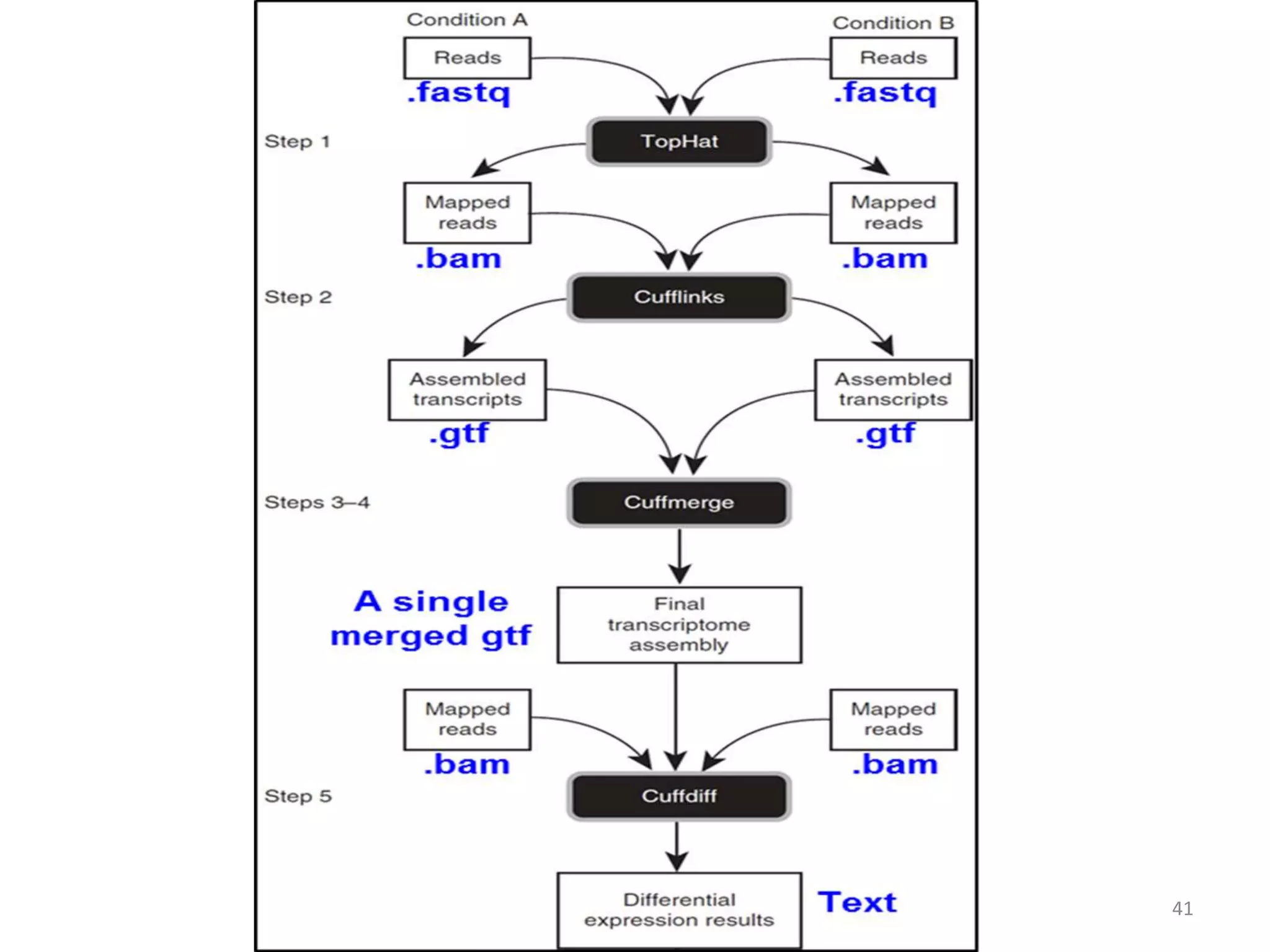
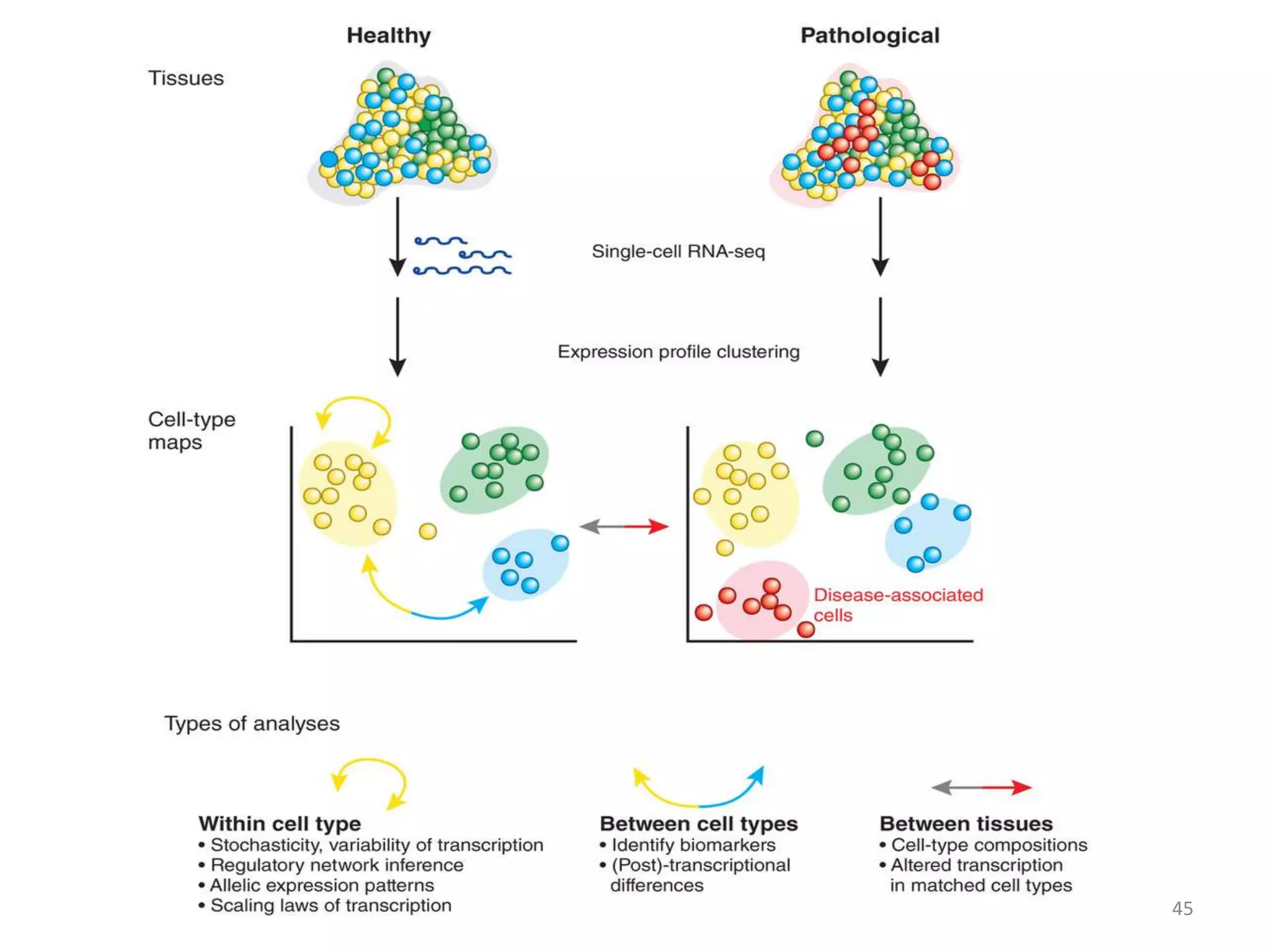
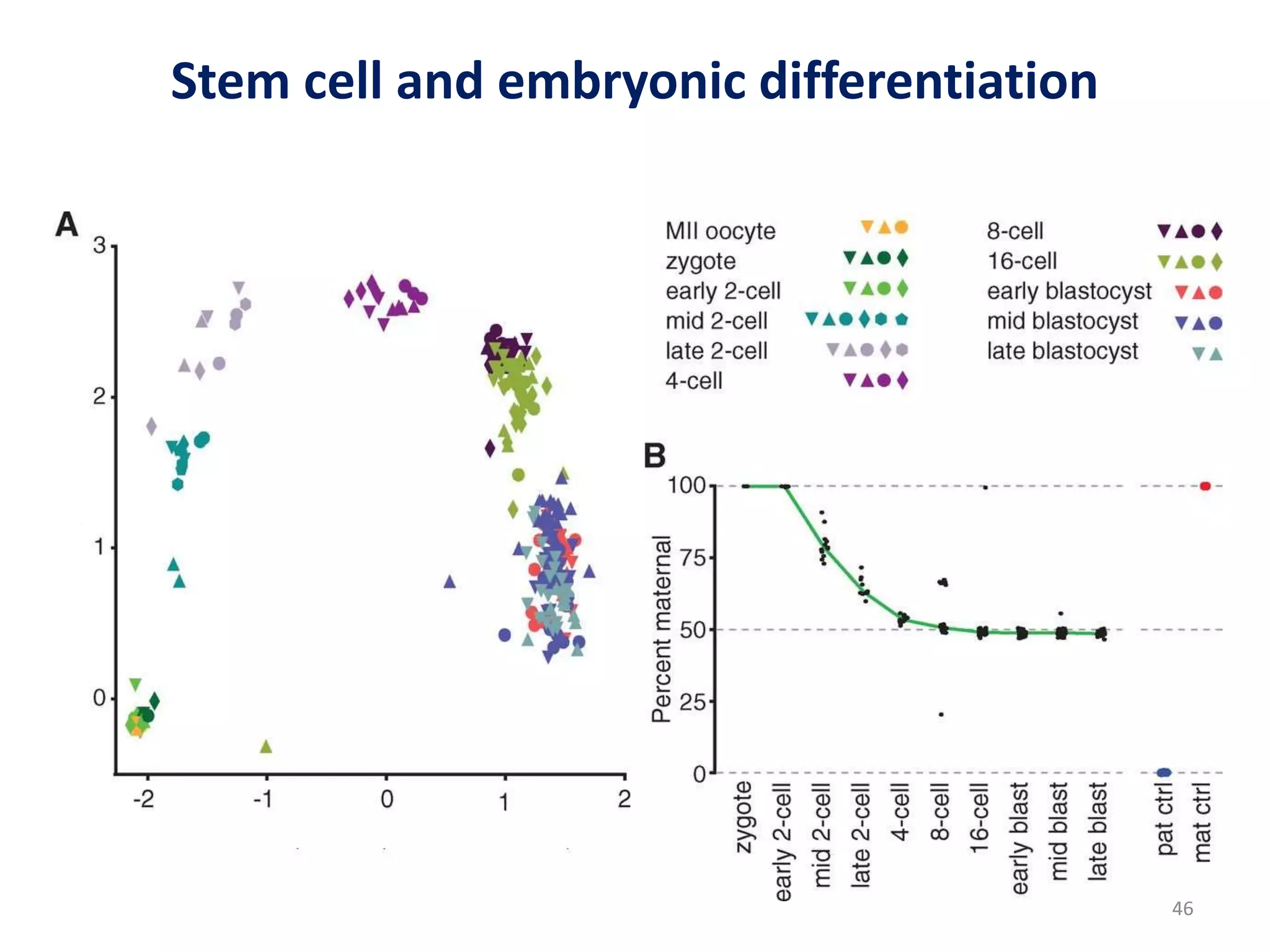
The document provides an overview of single-cell RNA sequencing (scRNA-seq), detailing its significance, methods of cell isolation, sequencing processes, and data analysis techniques. It highlights how scRNA-seq can reveal variations in gene expression and is particularly useful for studying rare cell populations. Additionally, it discusses various methodologies including fluorescence-activated cell sorting (FACS), micromanipulation, and applications in disease monitoring and stem cell research.