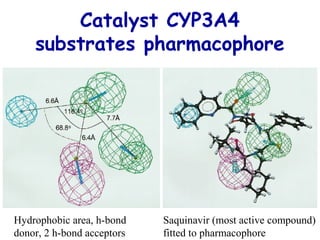
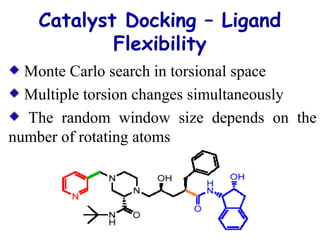
The document discusses quantitative structure-activity relationship (QSAR) methodologies, focusing on the mathematical relationships between biological activity and molecular properties for drug design. It details techniques like 3D-QSAR and software tools for predicting molecular interactions and activity, such as COMFA and VolSurf, while addressing assumptions, descriptor calculations, and the application of molecular fields. Furthermore, it highlights the challenges in superposition, molecular flexibility, and predictive accuracy in the context of cytochrome P450 substrates.