Download to read offline



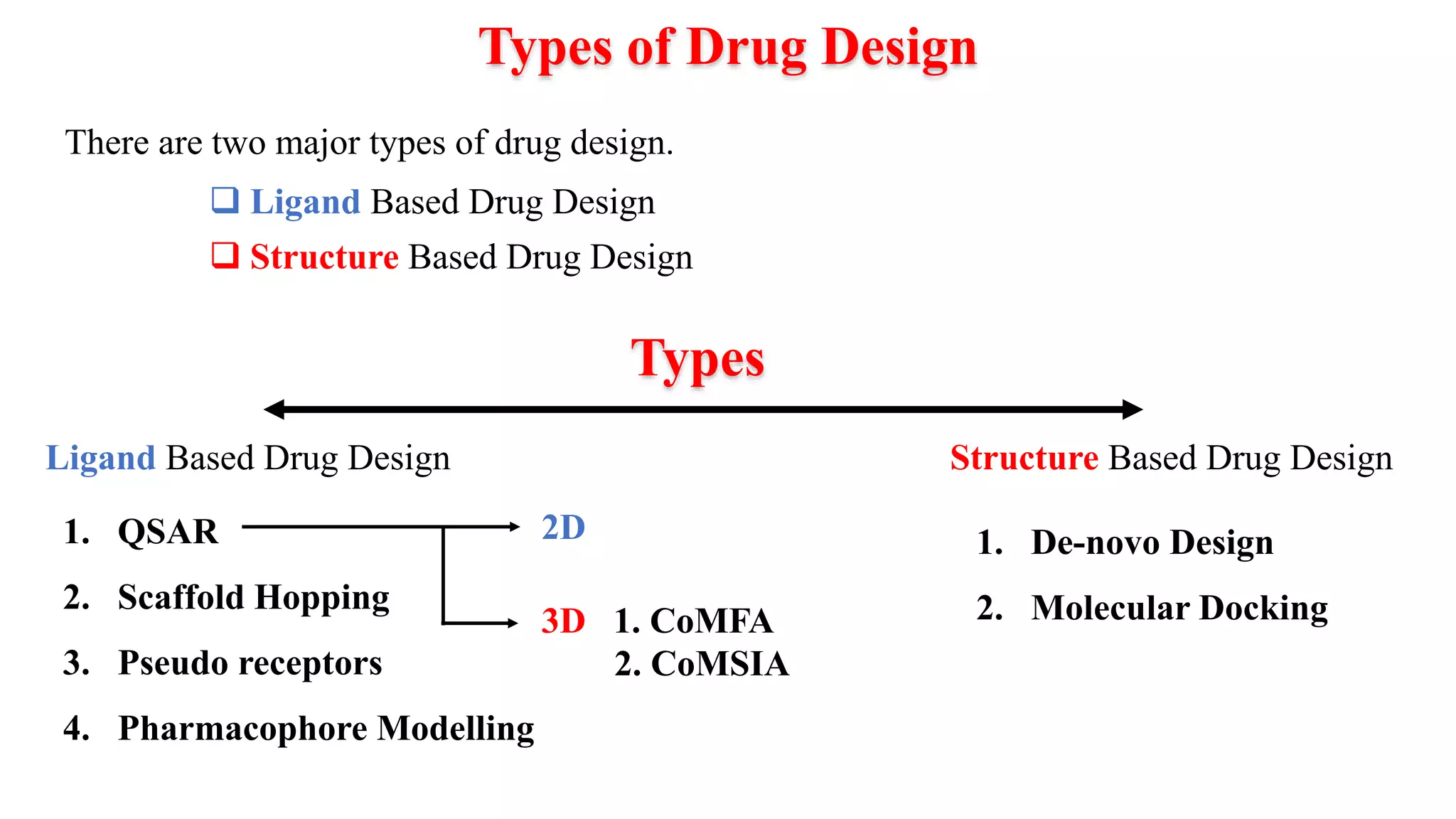
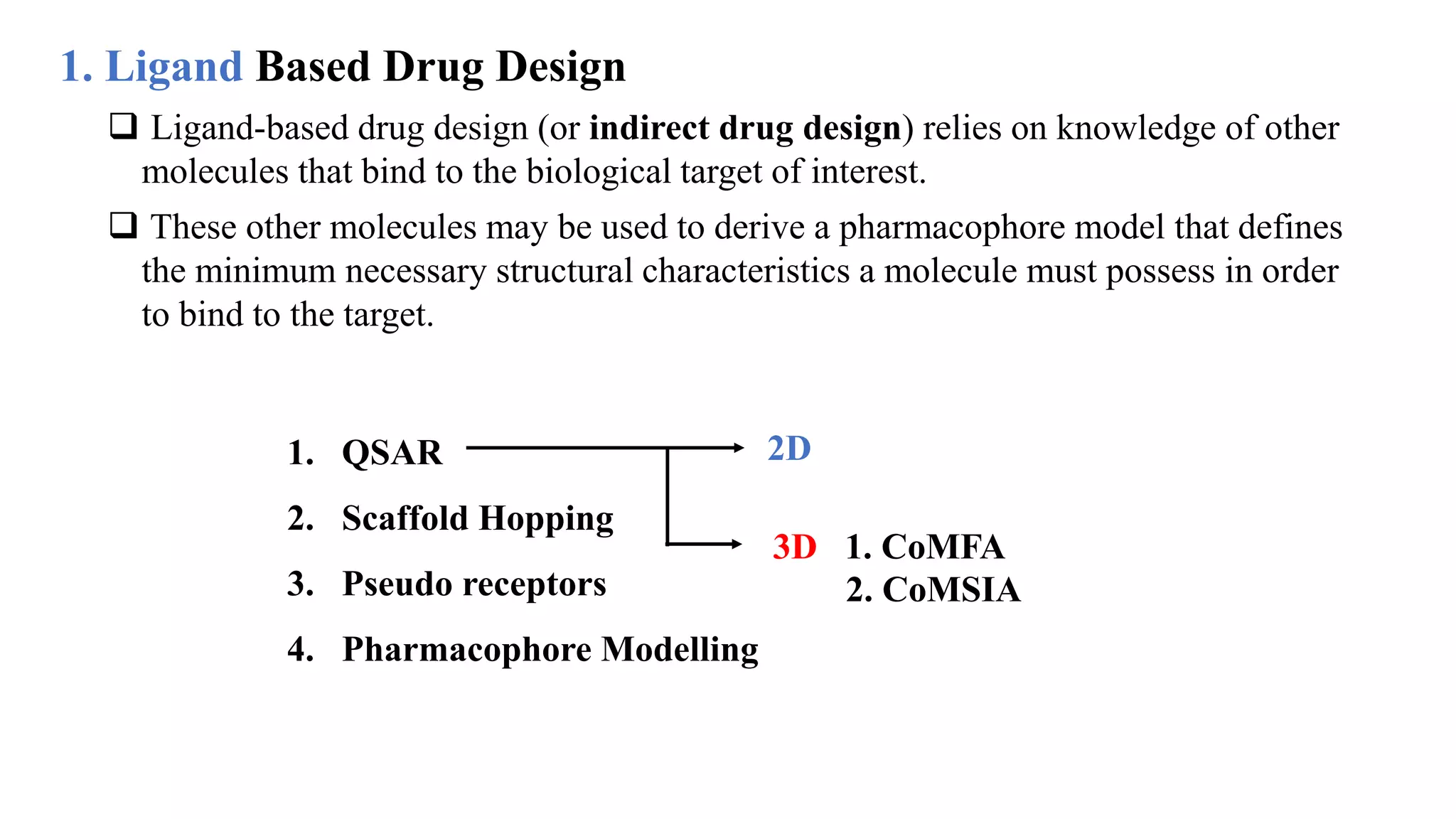

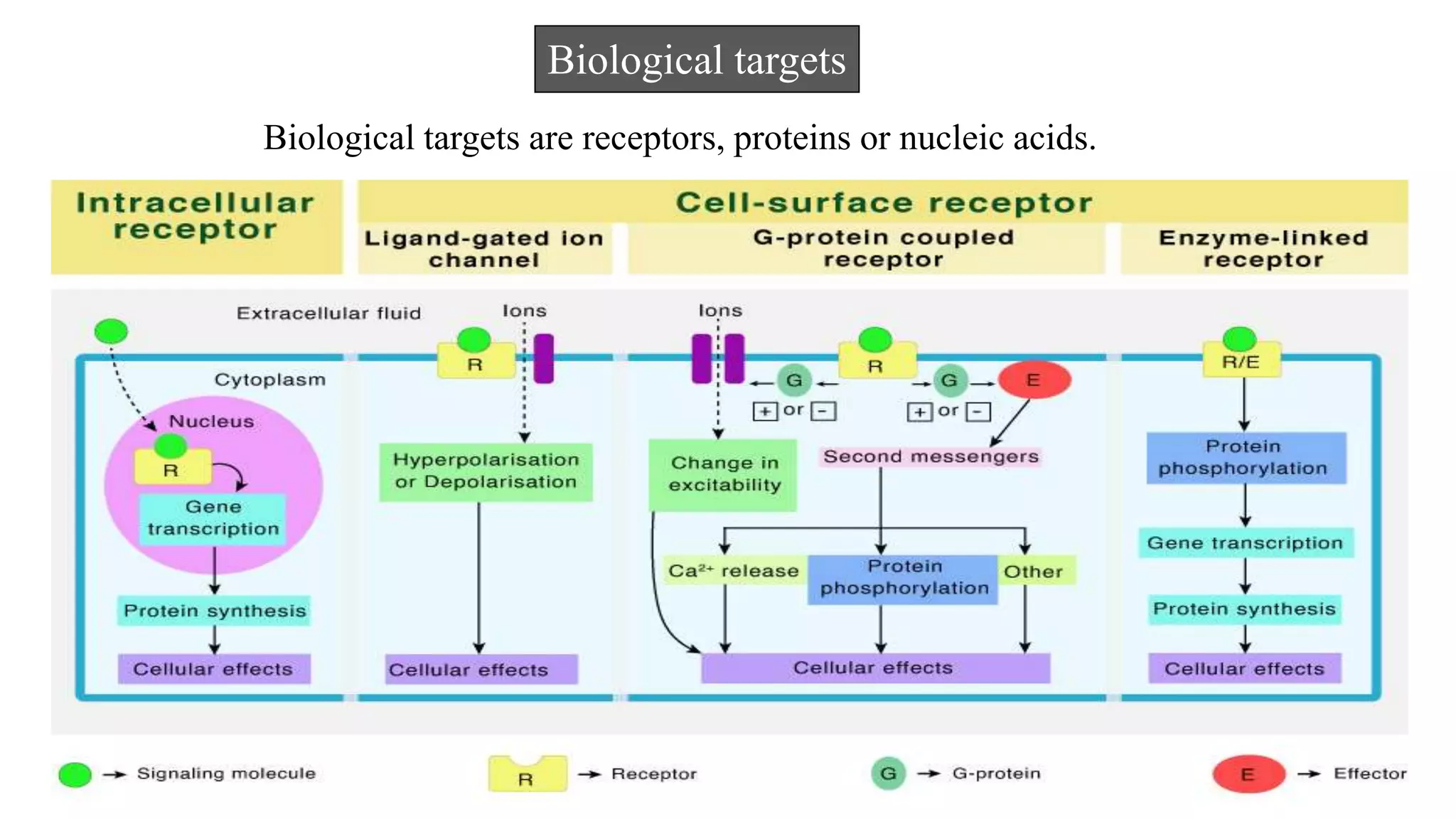


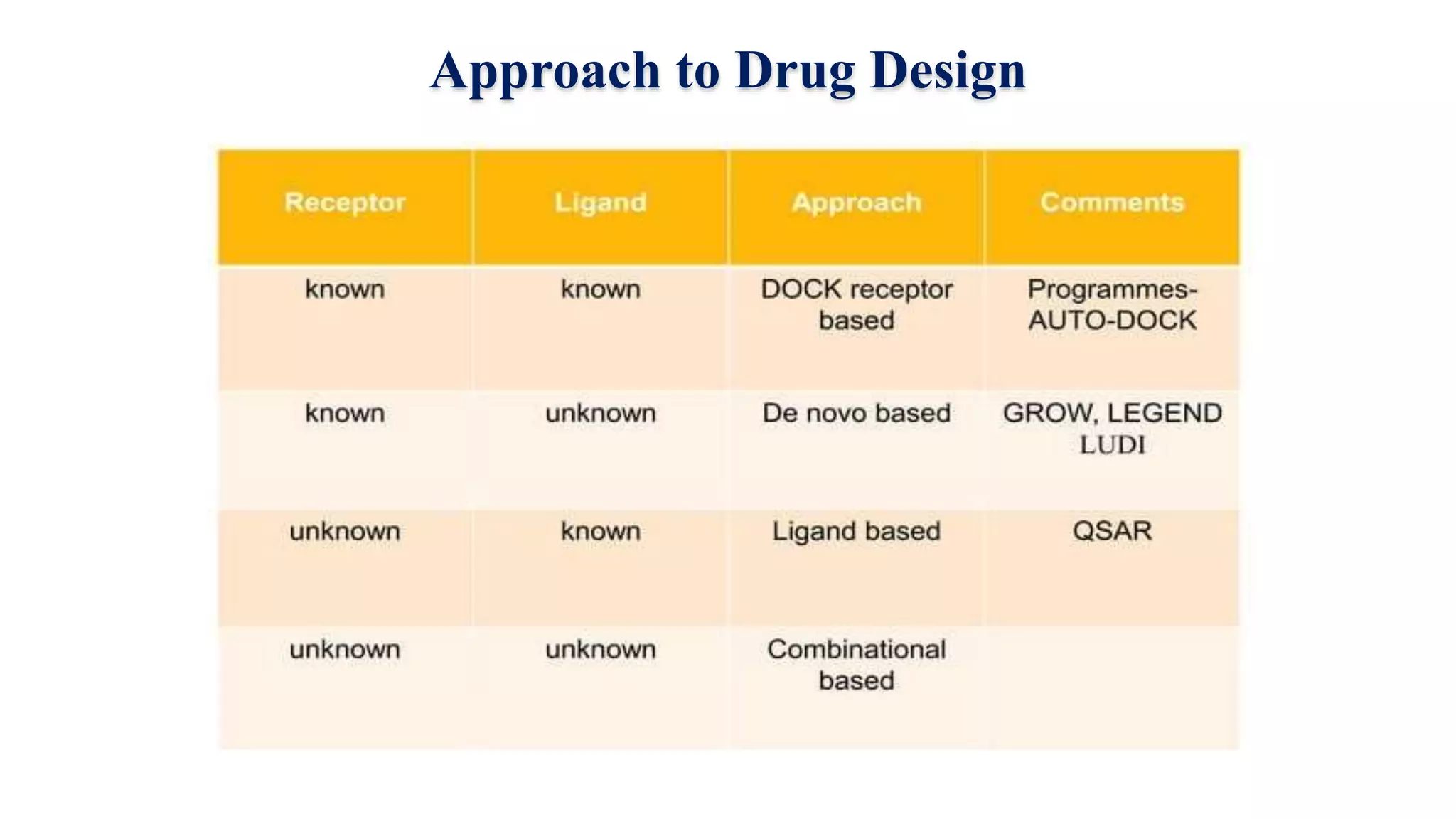



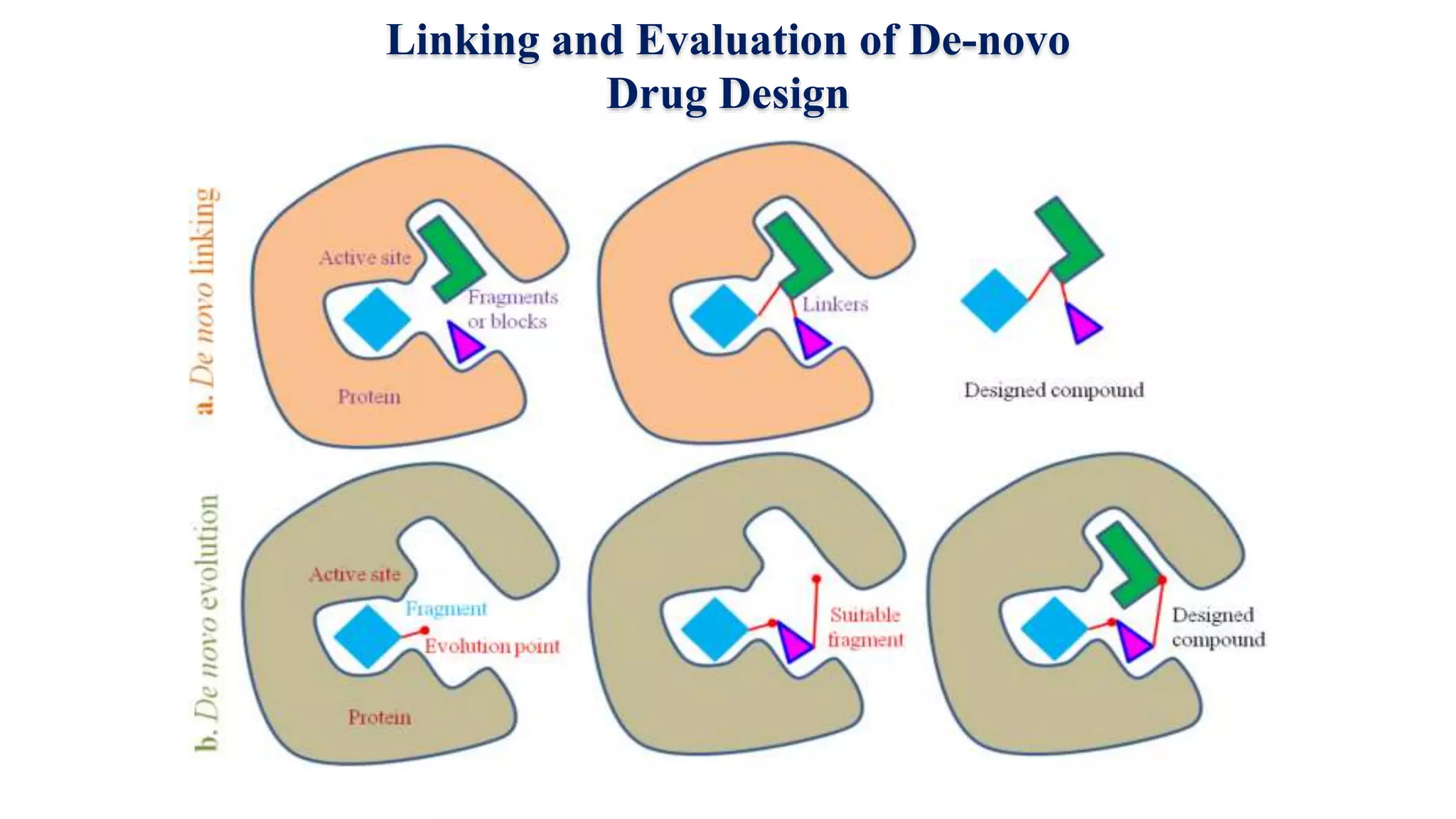
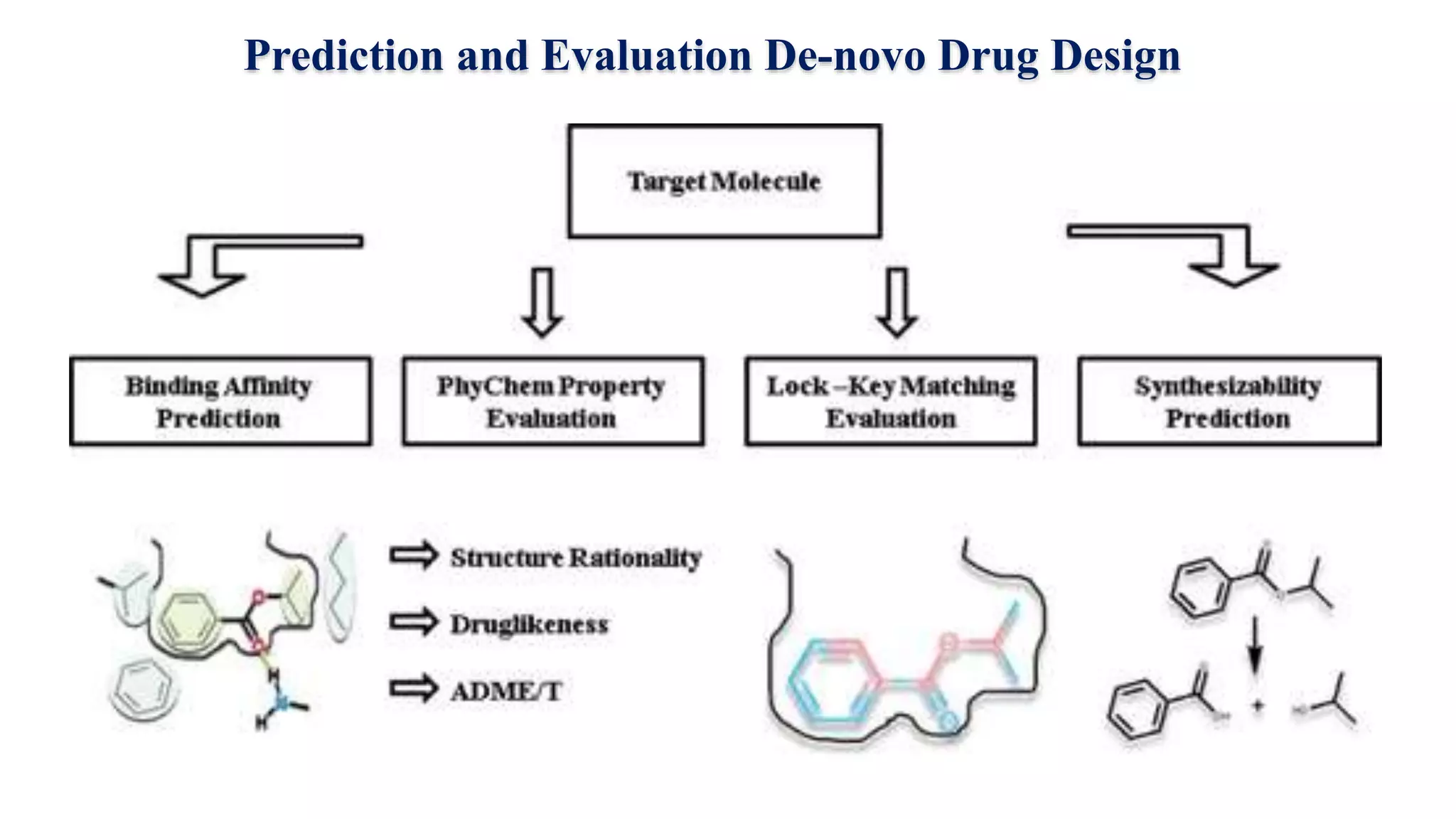












Hygia Institute of Pharmaceutical Education and Research provides information on drug design. There are two main types of drug design: ligand-based which relies on existing molecules that bind to the target, and structure-based which relies on the 3D structure of the target. De-novo drug design uses the 3D structure of the receptor to design new molecules and involves optimizing ligands to fit the receptor's active site properties. LUDI software aids de-novo design through identifying interaction sites in the receptor, fitting molecular fragments, and linking fragments together to form new drug candidates.























































































