









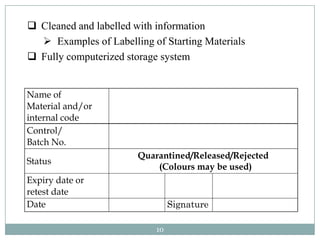






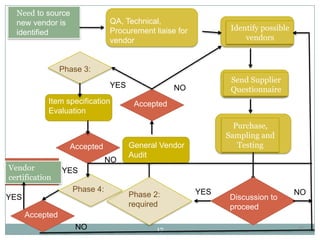
























The document discusses specifications for raw materials, finished products, and documentation. It defines specifications and their importance in ensuring quality. Specifications include tests, acceptance criteria, and procedures for materials and products. The document outlines specifications for raw material purchasing, storage, and testing as well as finished product testing, documentation, and storage. It discusses the relationship between specifications and regulations/standards.























































































