Downloaded 30 times













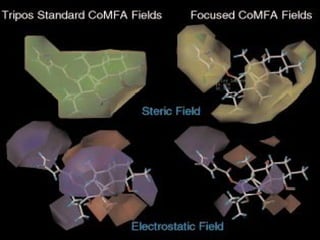






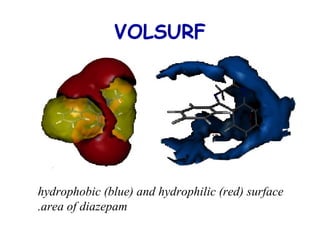




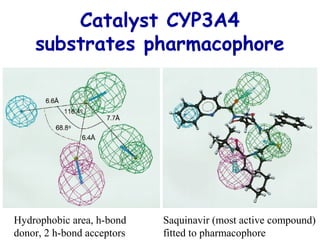
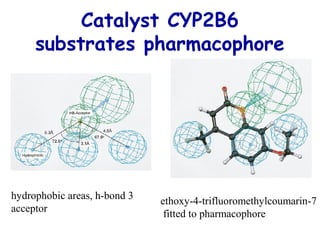

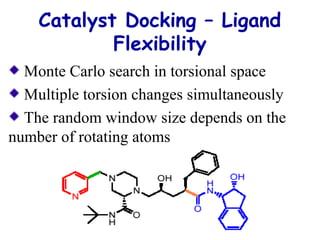







This document discusses quantitative structure-activity relationship (QSAR) modeling and 3D-QSAR techniques. It explains that QSAR aims to find consistent relationships between biological activity and molecular properties in order to predict activity of new compounds. It also describes several common 3D-QSAR software programs and techniques, including CoMFA, VolSurf, Catalyst, and DOCK, and provides examples of their applications to modeling various cytochrome P450 enzymes.











































































































