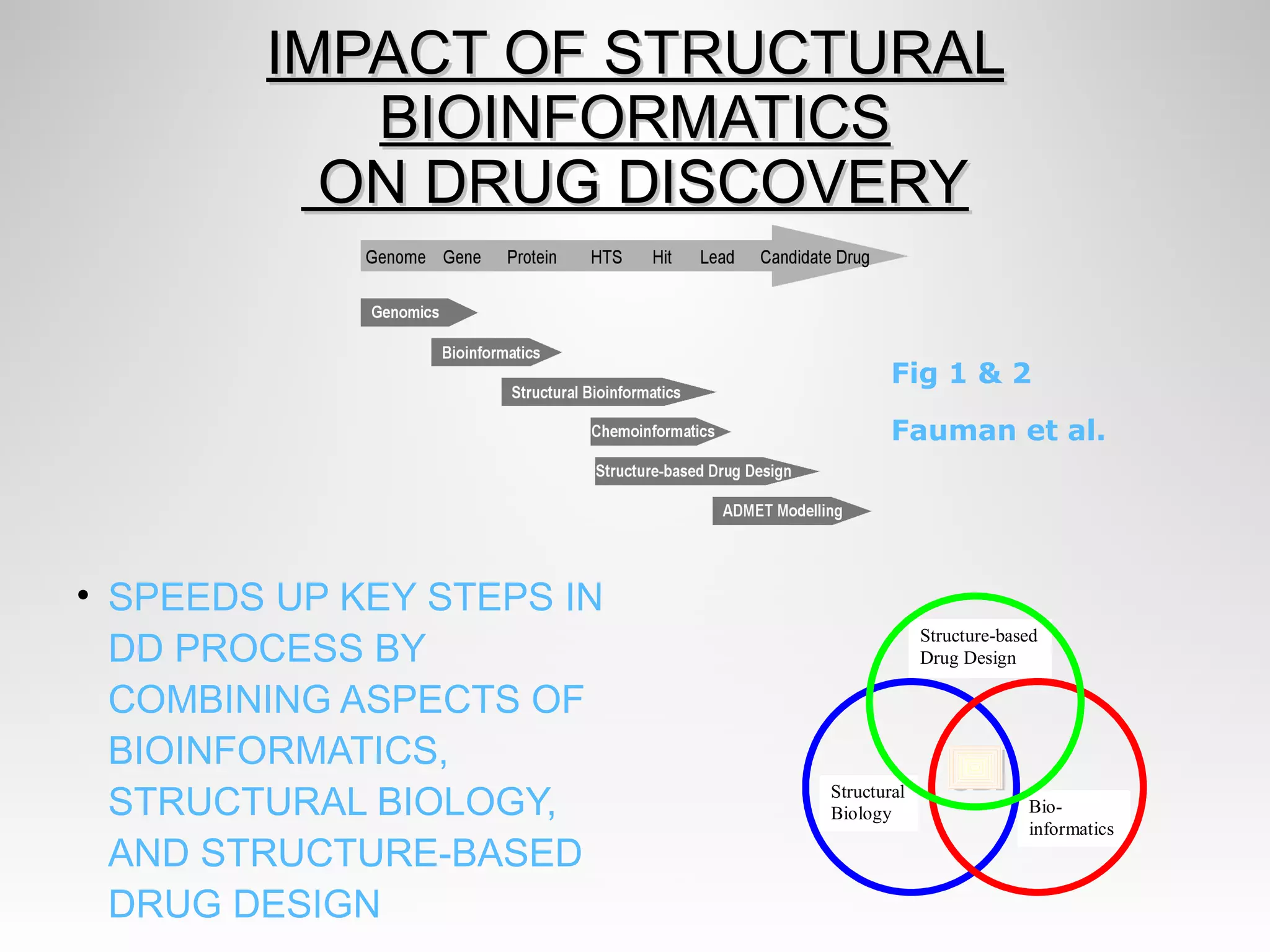
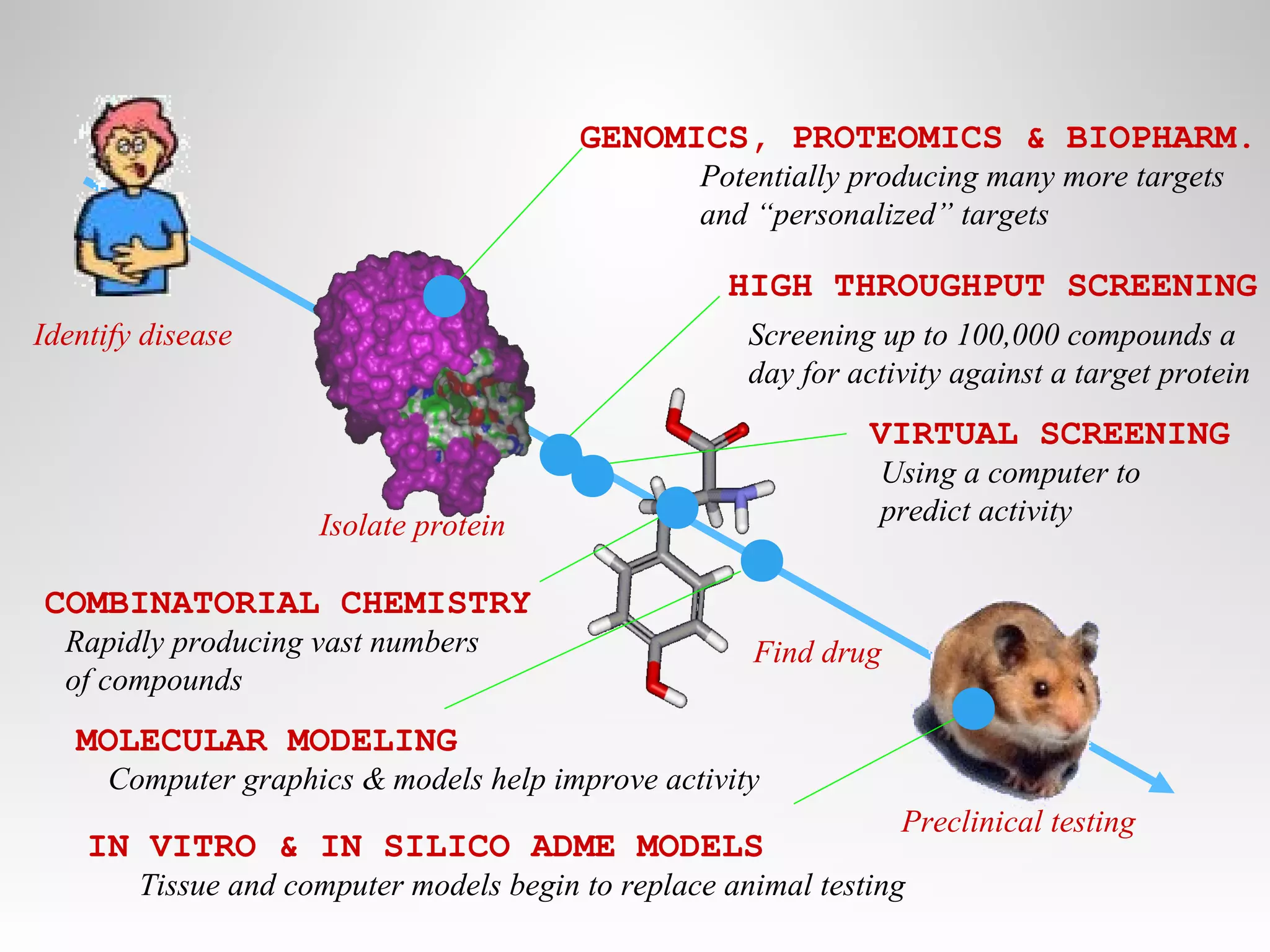
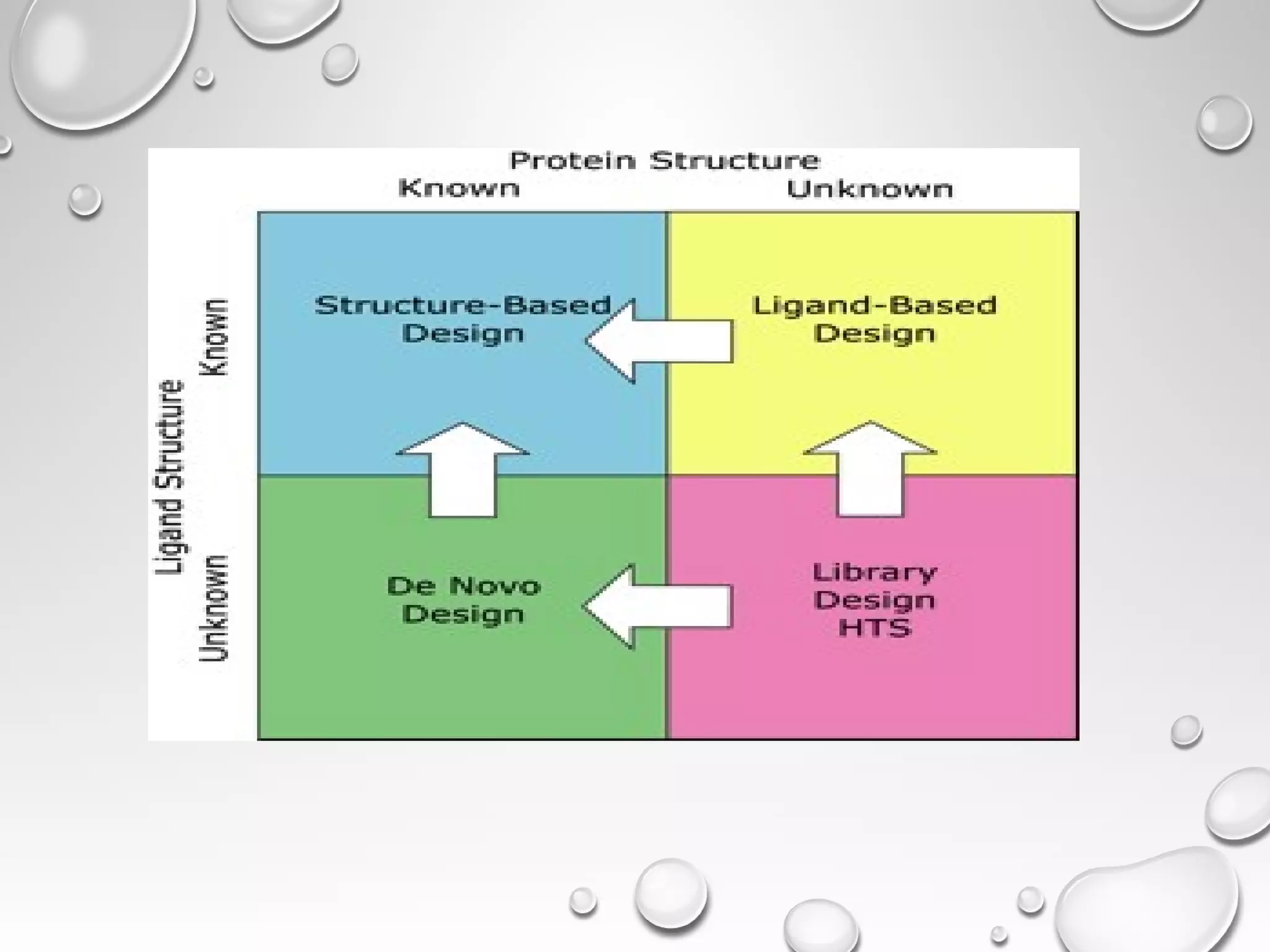
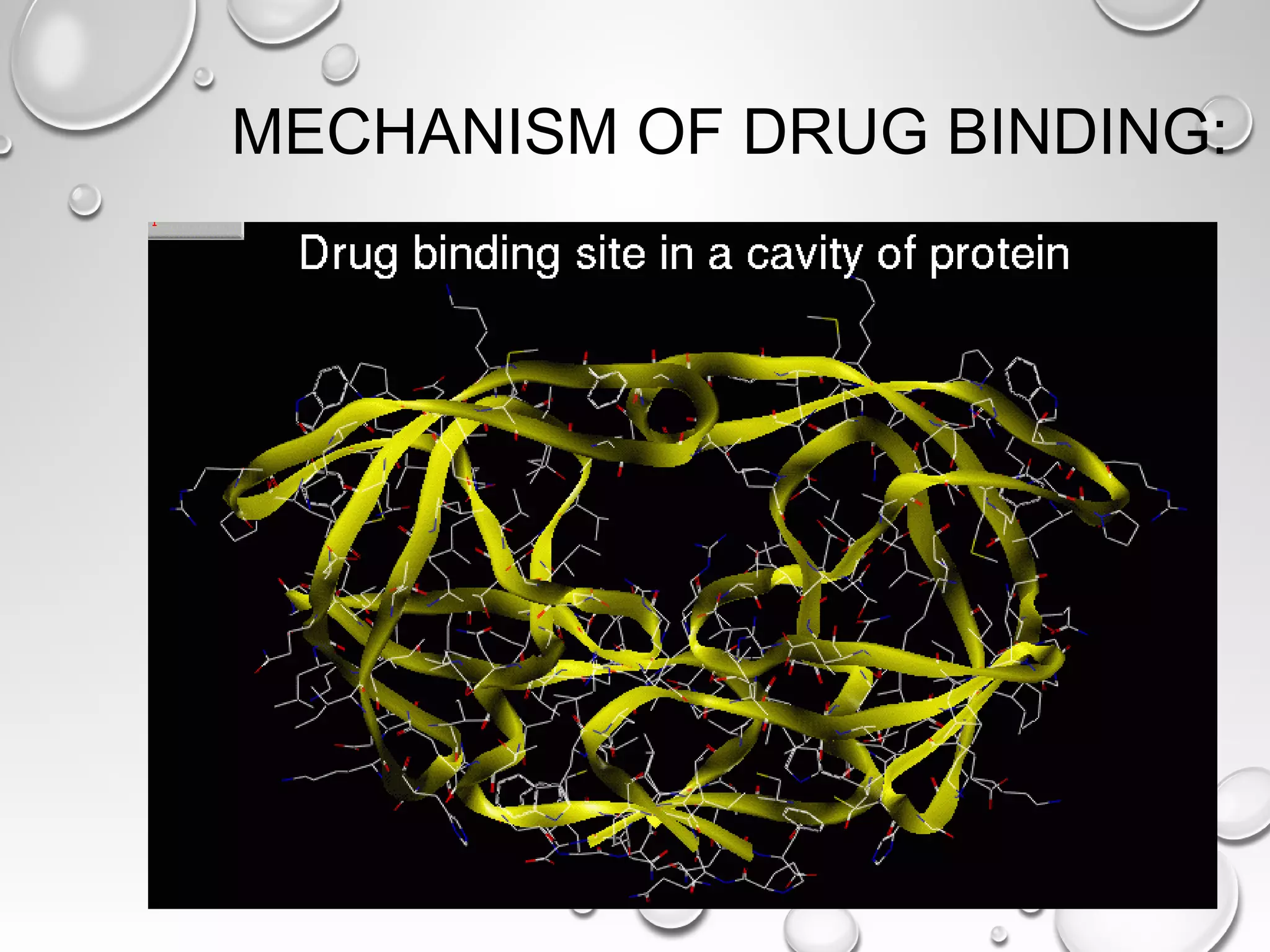
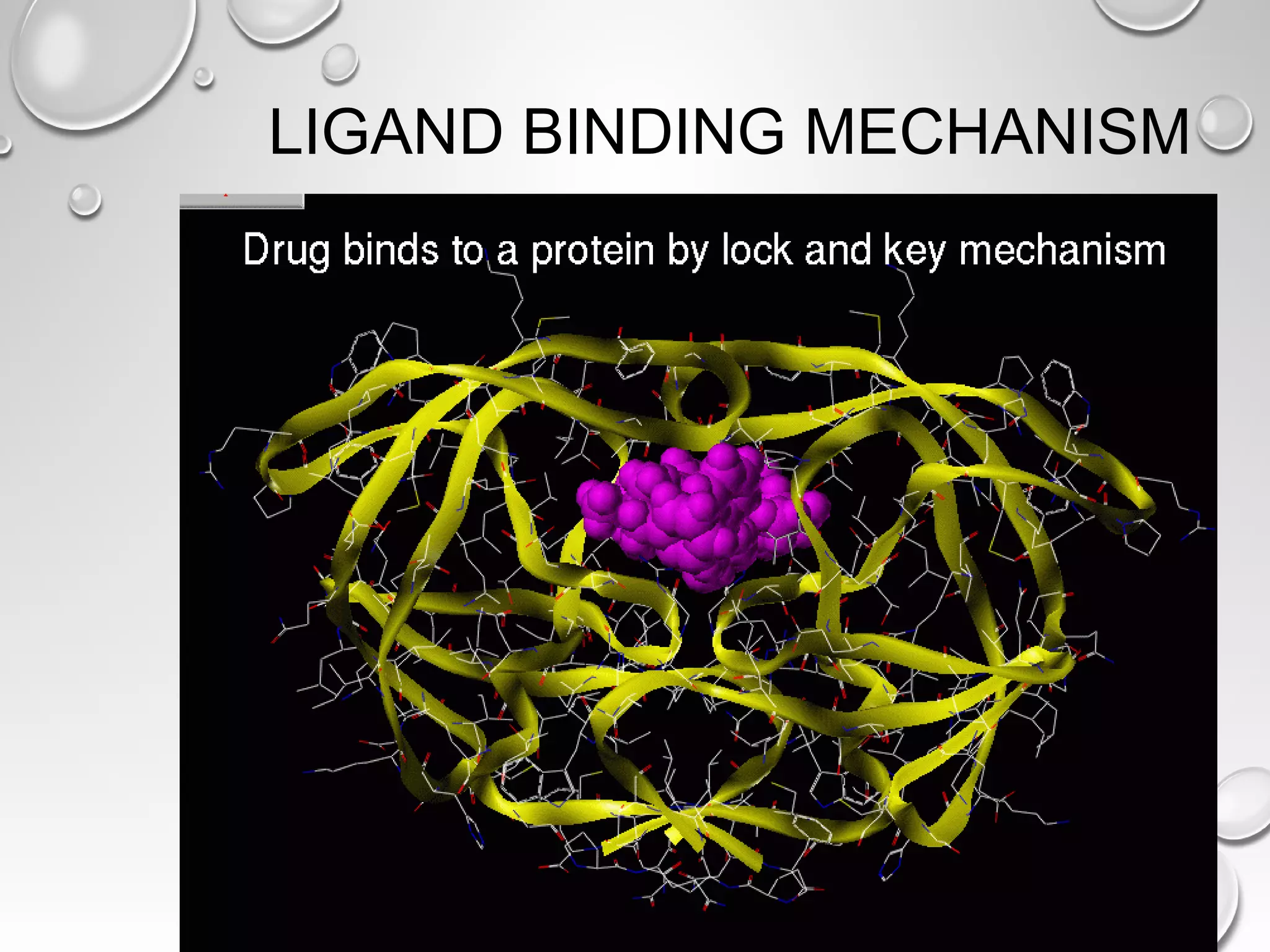
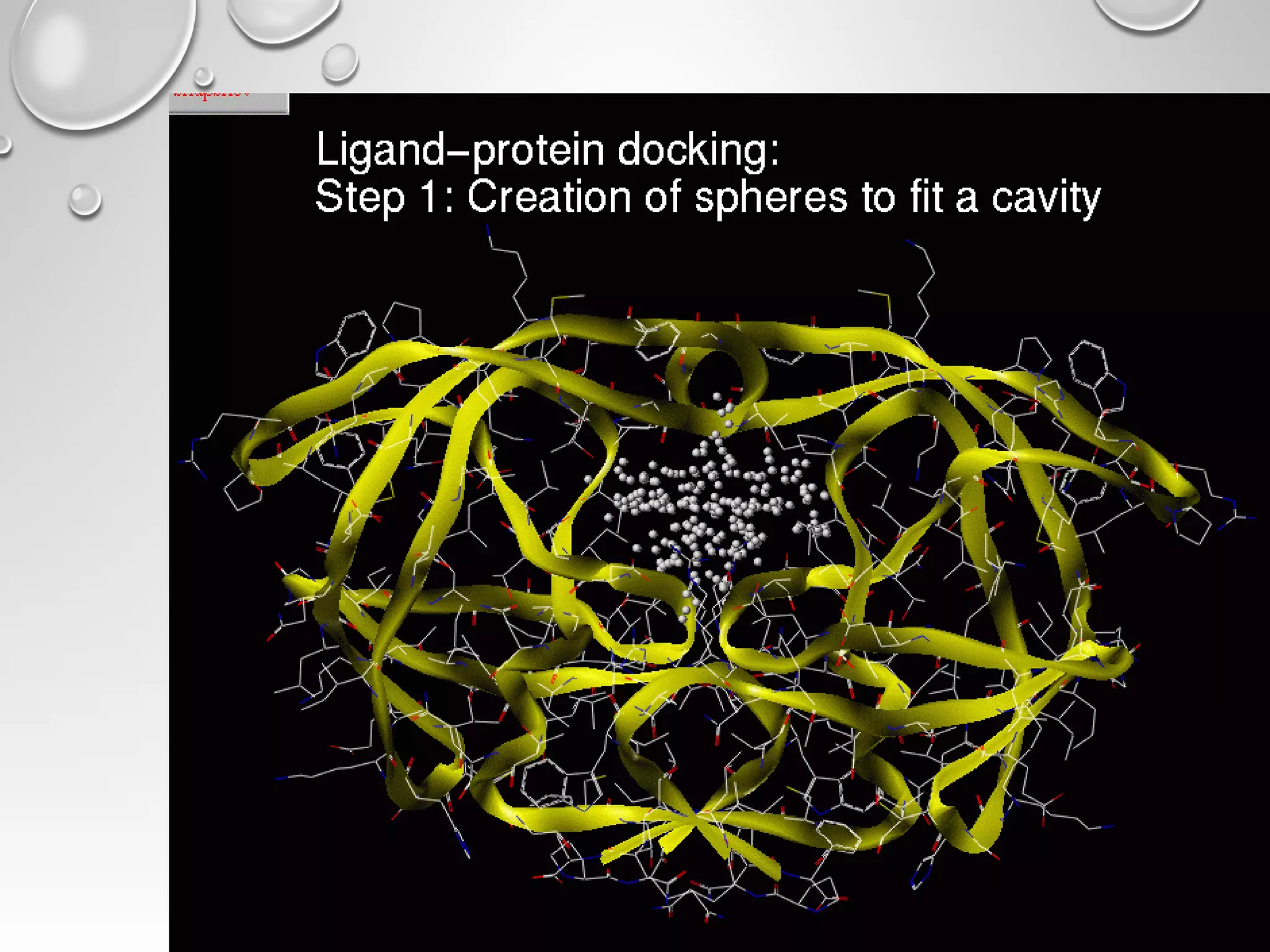
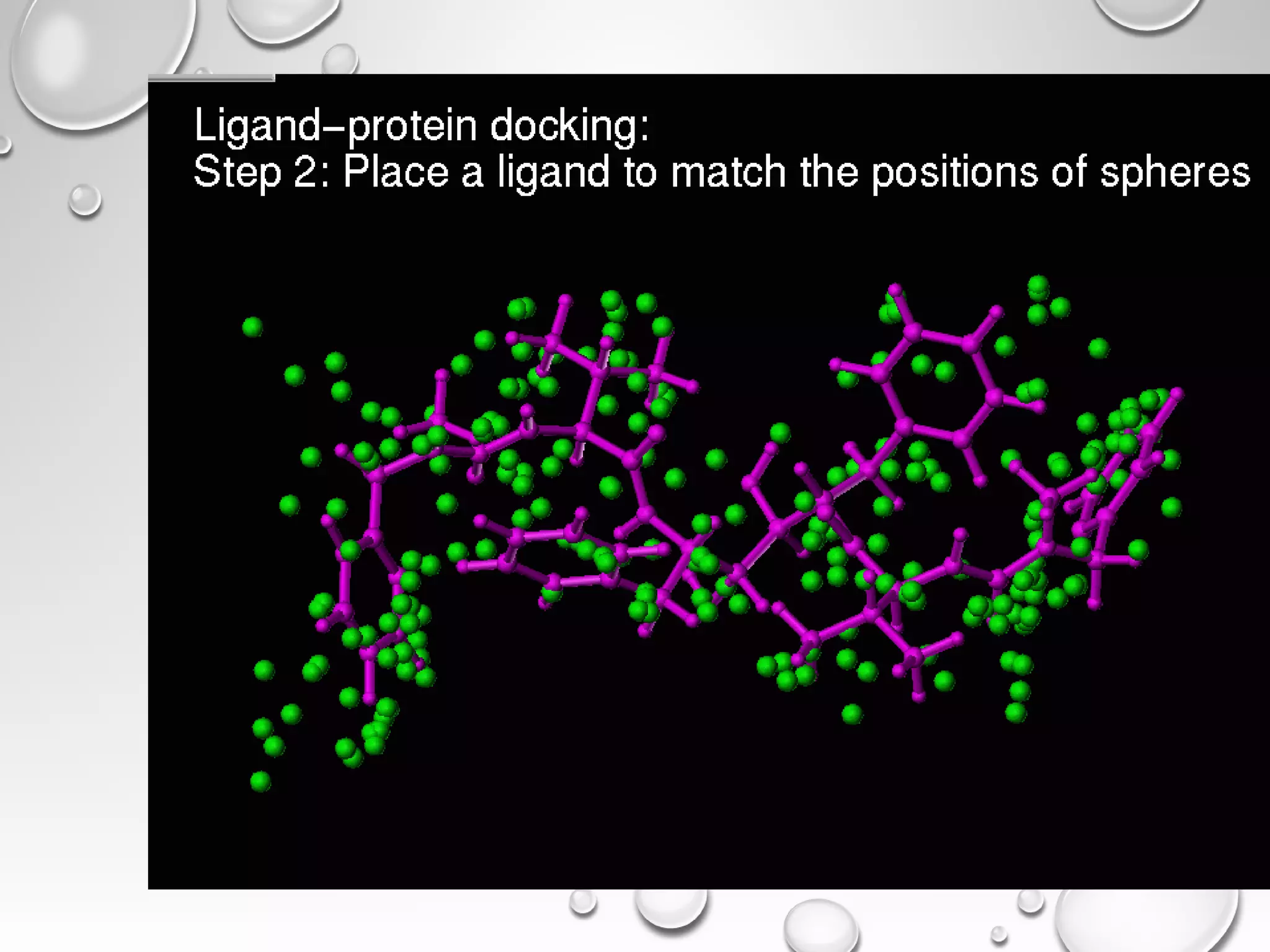
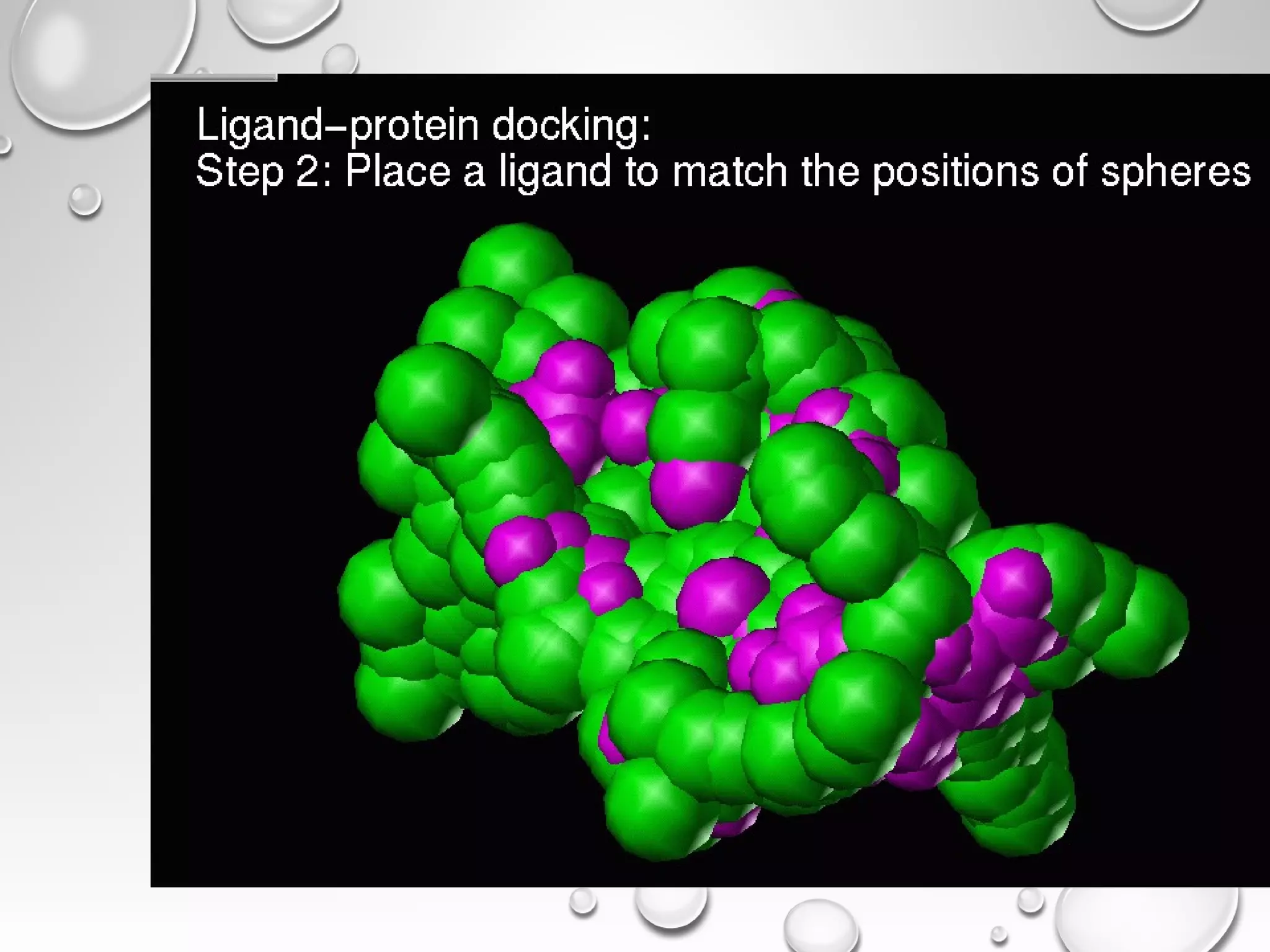
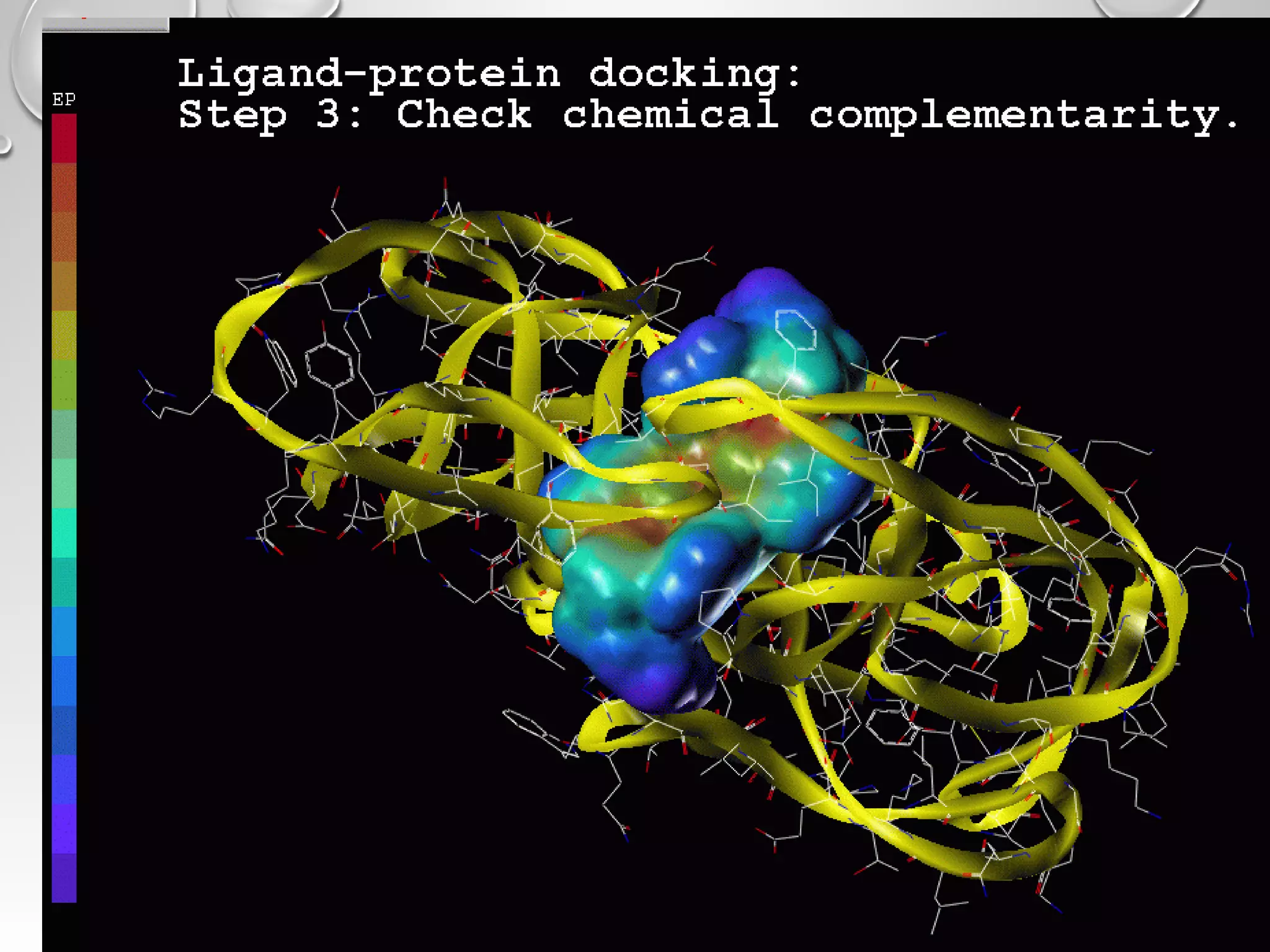
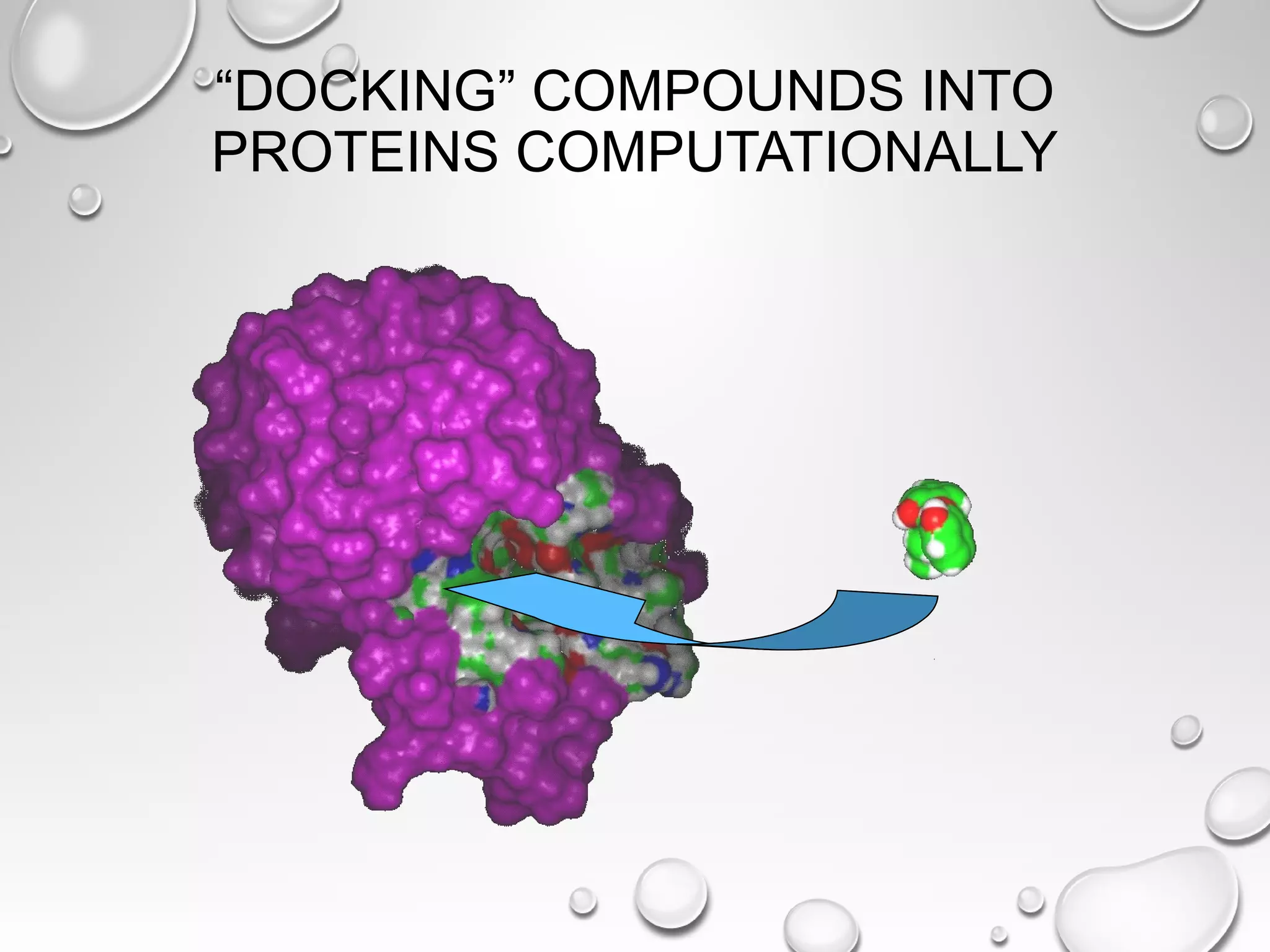
This document discusses computer aided drug design (CADD) and the drug discovery process. It describes how CADD uses computational methods to help identify and optimize lead compounds for drug development. The key steps in CADD and drug design include target identification, lead identification through screening libraries of molecules, lead optimization using techniques like docking to improve binding, and preclinical/clinical testing of potential drug candidates. CADD aims to accelerate drug discovery by reducing costs and experiments through in silico methods like structure-based drug design.




















































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)









