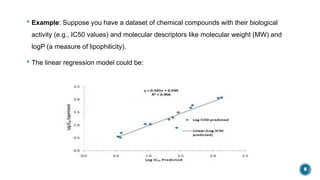
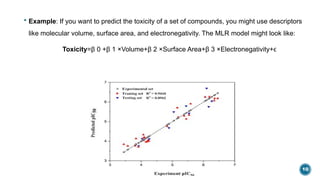
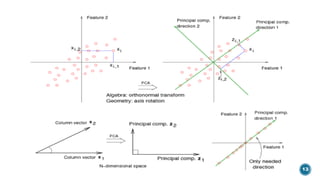
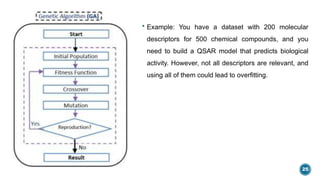
The document provides an overview of statistical methods used in quantitative structure–activity relationship (QSAR) modeling, crucial for correlating molecular structure with biological activity. It covers various statistical techniques such as linear regression, multiple linear regression, principal component analysis, and support vector machines, detailing their applications and advantages in QSAR. Additionally, it discusses the importance of cross-validation and the use of genetic algorithms for optimizing QSAR models.