Download as PPSX, PPTX
















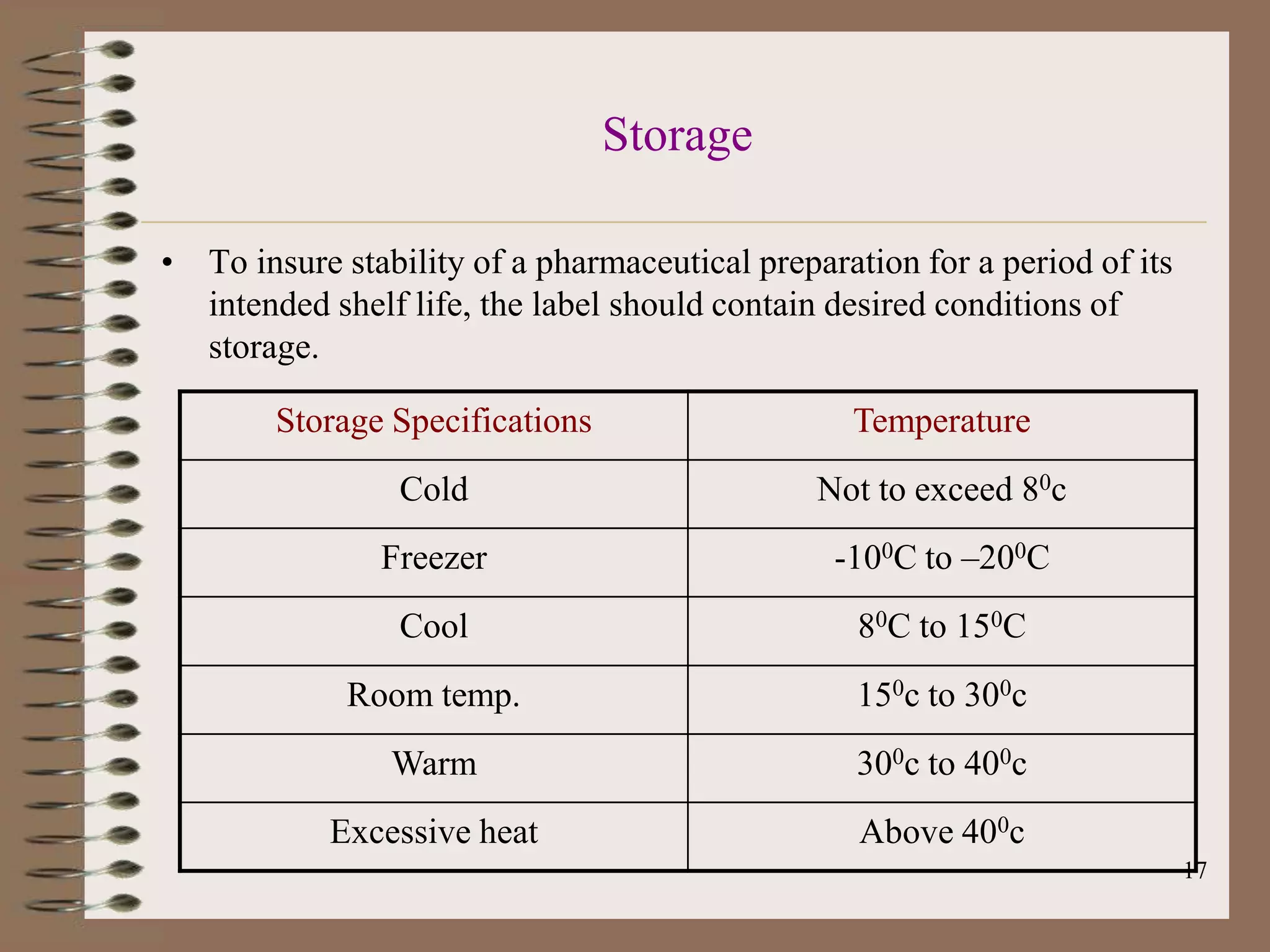











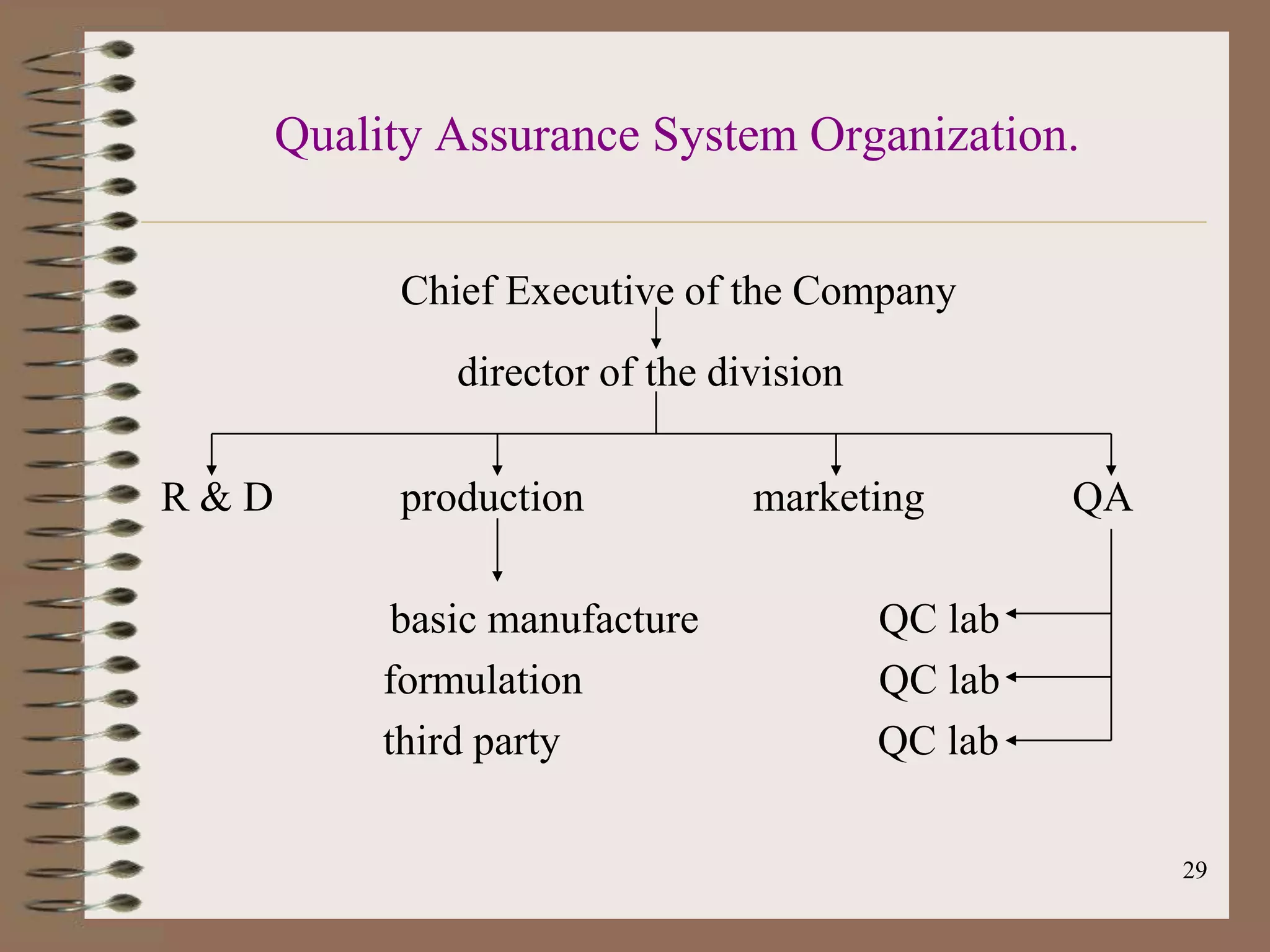
































The document discusses Good Manufacturing Practices (GMP) and Quality Assurance principles in pharmaceutical production, emphasizing the importance of quality at every manufacturing stage to ensure safety and efficacy. It outlines key aspects of GMP, including personnel qualifications, facility design, raw material handling, equipment maintenance, and documentation protocols. The document also highlights the functions of the Quality Assurance department, including batch production record reviews, complaint handling, and ensuring compliance with regulatory standards.











































































































