Downloaded 594 times






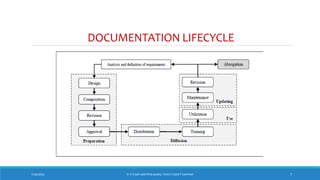










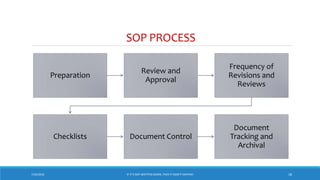











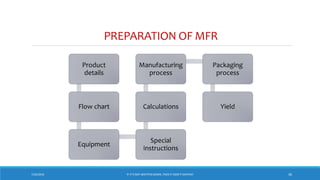











The document underscores the importance of pharmaceutical documentation as a cornerstone of quality management and GMP compliance, emphasizing that proper documentation minimizes risks and ensures that processes are clear and traceable. It outlines key objectives, types, and characteristics of effective documentation, along with guidelines for creating standard operating procedures (SOPs) and maintaining records such as master formula and batch records. The document also covers audit practices and specifications related to quality control in pharmaceutical production.




































![supriya.k_ppt[1].pptx documentation for QA and QC](https://cdn.slidesharecdn.com/ss_thumbnails/supriya-250809112814-eb4e6f1f-thumbnail.jpg?width=640&height=640&fit=bounds)
































![Hypothalamus short notes on location, function and disorders by Dr. Neha [PT]...](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124142231-2b48143d-thumbnail.jpg?width=640&height=640&fit=bounds)


![Cells and Organs of immune system [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/cellsandorgansofimmunesystemautosaved-260123152717-ea0cb261-thumbnail.jpg?width=640&height=640&fit=bounds)






