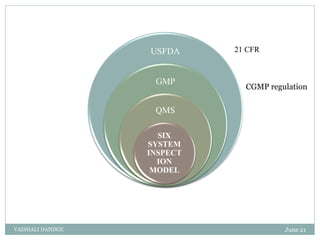
The document discusses quality management systems and the six system inspection model used by the FDA to ensure compliance with cGMP regulations. It describes each of the six systems - quality system, production system, facilities and equipment system, laboratory control system, materials system, and packaging and labeling system. For each system, it provides an overview and lists the relevant cGMP subparts that govern inspections of that system. The goal is to help pharmaceutical manufacturers implement quality systems to meet FDA requirements.