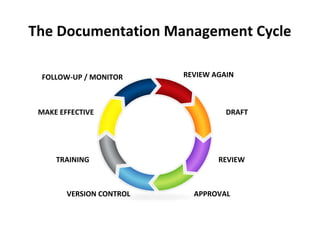
This document discusses good documentation practices for GMP compliance. It defines what documentation is, outlines the types of documents required by GMP such as batch records and SOPs. It explains the importance of documentation for meeting legal requirements, business needs, and enabling good decision making. It provides tips for writing good documentation including structure, approvals, version control, and retention. Overall it emphasizes that documentation is critical to demonstrate regulatory compliance and quality.











































![Observations on poor
documentation practices
Use of signature stamp
• US FDA Warning Letter UCM075960 to Scott A. Spiro, MD, 28-Jun-06. [16]
• US FDA Warning Letter UCM066113 to Medtronic, Inc., DEC 2 1997. [17]
Obscured original data
• US FDA Warning Letter UCM076246 to Gynetics Medical Products NV, JAN
16 2007[19]
Use of pencil
Inaccurate records
• US FDA Warning Letter 320-01-02 to SOL Pharmaceuticals Limited, NOV
21, 2000[18]
Hand changes not dated
• Form FDA 483 issued to L. Perrigo Co., NOV 7, 2008. [20]](https://image.slidesharecdn.com/gmpdocumentationpractise-dr-amsavel-121029051741-phpapp01/85/Good-Documentation-Pactise-dr-amsavel-45-320.jpg)




























































![supriya.k_ppt[1].pptx documentation for QA and QC](https://cdn.slidesharecdn.com/ss_thumbnails/supriya-250809112814-eb4e6f1f-thumbnail.jpg?width=640&height=640&fit=bounds)



























![5G Explained! A High Level Overview [Introduction]](https://cdn.slidesharecdn.com/ss_thumbnails/5gexplainedahighleveloverview-260119165306-cc137a3e-thumbnail.jpg?width=640&height=640&fit=bounds)






