I. This document outlines the key aspects of Good Manufacturing Practices (GMP) and cGMP, including a timeline of GMP development, requirements for personnel, premises, equipment, standard operating procedures, validation processes, and documentation such as batch records.
II. It defines GMP as ensuring consistent and controlled production of products according to quality standards. cGMP requirements include qualified personnel, designed facilities and equipment, and documented procedures for manufacturing, testing, and record keeping.
III. The document provides details on specific GMP rules for premises, equipment, personnel, operations, warehousing, validation, and labeling. Adherence to GMP aims to minimize errors and ensure uniform, high quality batches of pharmaceutical products.
CONTENT
Definition
Differencebetween GMP & cGMP
Time line of GMP
Personnel
Premises
Equipment
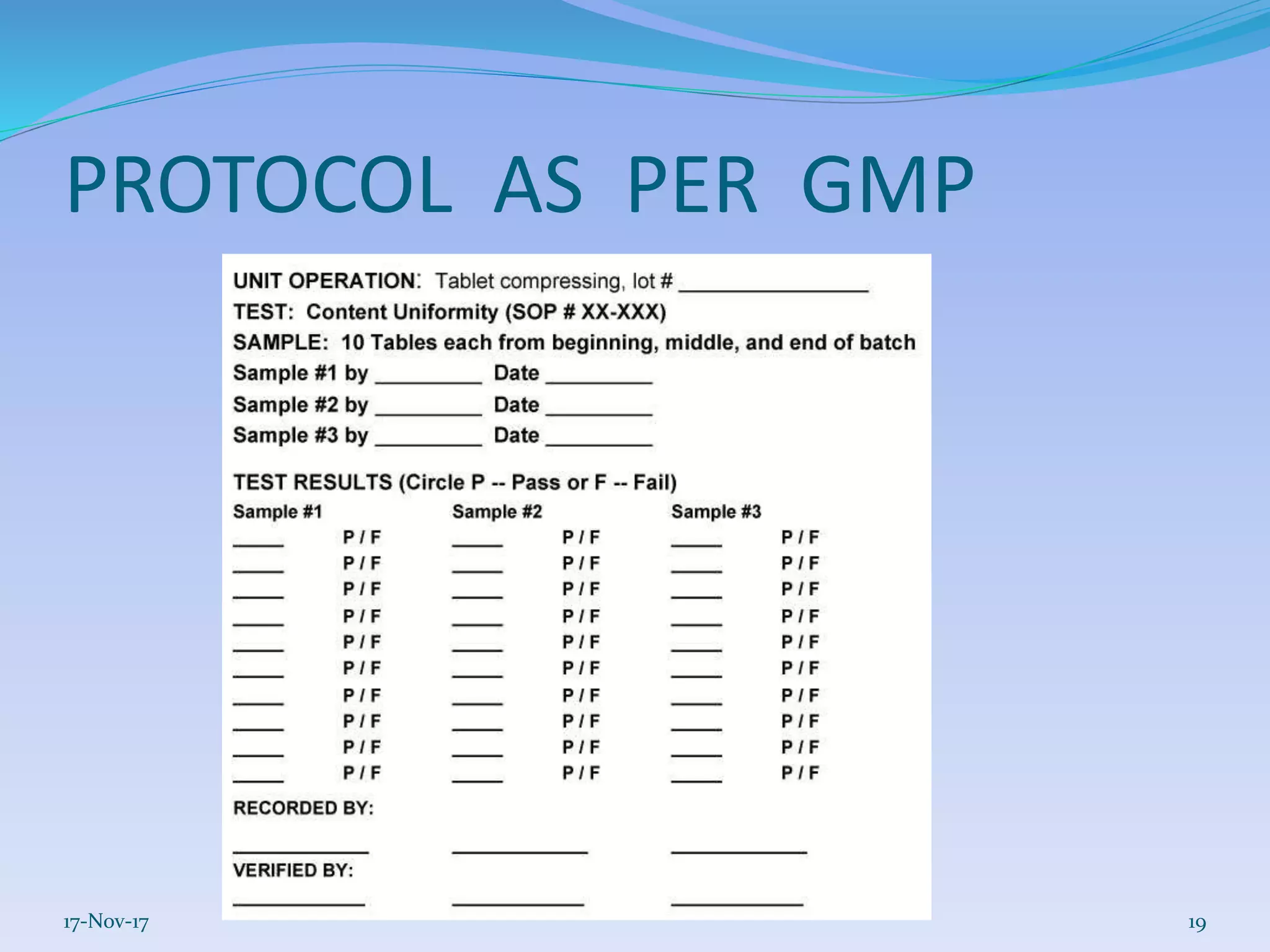
SOP(Standard Operating Process)
Master Formula Record and Batch Manufacturing Record
Validation and Validation Process & Protocols
17-Nov-17 2
3.
GMP:-
That part ofQA which ensures that
products are consistently produced and
controlled to the quality standards as per the
specifications.
17-Nov-17 3
•1902 - Developmentof the Biologic Control Act
•1906 - Development of the Pure Food and Drug Act
•1938 - Federal Food, Drug and Cosmetic Act
•1941 - Initiation of GMP
•1944 - Development of Public Health Services Act
•1962 - Kefauver-Harris Drug Amendments released
•1963 - Establishment of GMPs for Drugs
•1975 - CGMPs for Blood and Components Final Rule
•1976 - Medical Device Amendments
•1978 - cGMPs for Drugs and Medical Devices
•1979 - GLPs Final Rule
•1980 - Infant Formula Act is passed
17-Nov-17 5
Geography, climate, and economic factors
Neighbours
a) What can happen?
Premises must be located to minimize risks of cross-contamination,
e.g. not located next to a malting factory with high airborne levels of
yeast
Pollution/effluent control
17-Nov-17 10
11.
Minimize risks oferrors
Permit effective cleaning
Permit effective maintenance
Avoid cross-contamination, build-up of dirt and dust
Maximum protection against entry of insects, birds
and animals
Separate facilities for other products such as some
antibiotics, hormones, cytotoxic substances
17-Nov-17 11
12.
Eating ,Drinking, Smoking Should not be allowed in
the Production area.
17-Nov-17 12
13.
1. Measures shouldbe taken to prevent cross-
contamination
2. Dust control measures (including extraction of dust
and air)
3. No areas for dust accumulation
4. Easily cleanable surfaces
5. Proper air supply
6. Use of HEPA filter’s
17-Nov-17 13
14.
Equipment shallbe located, designed, constructed,
adapted and maintained to suit the operation to be
carried out.
Should be made of non reactive material, such as High
grade of steel(316,302)
Equipment should be-
a) Calibrated
b)Checked
c)labeled
d)Sterilized
17-Nov-17 14
15.
Written procedures
•hygiene, health and clothing practices
• waste disposal
Implementation and training
Practices not permitted
a)eating, smoking
b) unhygienic practices
17-Nov-17 15
16.
There shallbe written Standard Operating Procedure for
each operation
It include-
a)For Equipments
b)For sampling
c)For Testing
d)For Process
f)For Packaging
17-Nov-17 16
17.
An Inventoryshould be maintained for Raw materials
to be used at any stage of manufacture
Records should be maintain as per Schedule U
Should be purchased from approved sources
Must be checked by QC department on recipt
Should be labeled.
17-Nov-17 17
18.
There shallbe MFR relating to all manufacturing procedures for
each product and batch size to be manufacture
It should include-
i)The name of the product
ii)Quantity, of all starting materials to be used
iii)A statement of the expected final yield with acceptable
limits.
iv) Principal equipment to be used
v) Detail stepwise processing instructions and the time taken for
each step
vi)Any special precautions
vii)Packing details and Specimen labels
17-Nov-17 18
There shallbe Batch processing record for each product.
During Manufacturing or Processing the following
information shall be recorded
It include-
The name of the product
The number of Batch being manufactured
Dates and time of commencement of batch and
completion
Initials of operator
Amount of Product obtained
17-Nov-17 21
22.
I. Warehousing areashould be designed and adapted
to ensure good storage conditions.
II. Should be Clean, dry and maintained with
acceptable temperature limits.
III. Should have appropriate house-keeping and
rodents, pests and vermin control.
IV. Separate sampling area for active raw material and
exciepients.
V. Every Material stored should be labeled properly.
Fire Prevention
17-Nov-17 22
23.
Essential partof GMP
Necessary to achieve the intended results
A written record is prepared summarizing recorded result and
conclusions shall be prepared ,documented and maintained
Should be necessary when-
a)Any new master formula or method of
preparation is adopted
b)For critical process
c)any changes in the equipment, or when
using a new equipment, it is first validated
to demonstrate its consistentency of
required quality
17-Nov-17 23
All containersand equipment should bear labels
Different color coded labels should be used to indicate
the status of a product(for example under
test,approved,passed,rejected)
The Printing should be done in bright colors
The label should contain all the prescribed details
about the product.
17-Nov-17 25