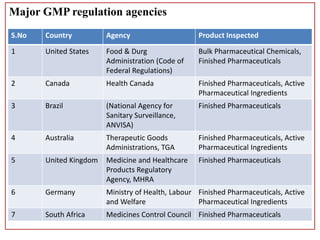
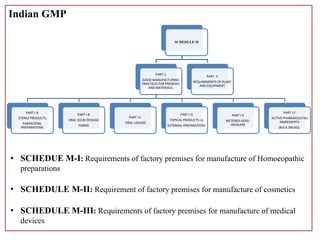
This document defines Good Manufacturing Practice (GMP) and outlines its history and key regulations. GMP is a set of guidelines for ensuring products are consistently produced and controlled according to quality standards for their intended use. Major developments include the 1962 Thalidomide tragedy leading to GMP regulations, and guidelines published by organizations like WHO, FDA, and other national regulatory agencies. The document describes key aspects of GMP including facilities, equipment, production processes, packaging and labeling, quality control, and record keeping.





















































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)













