Downloaded 253 times









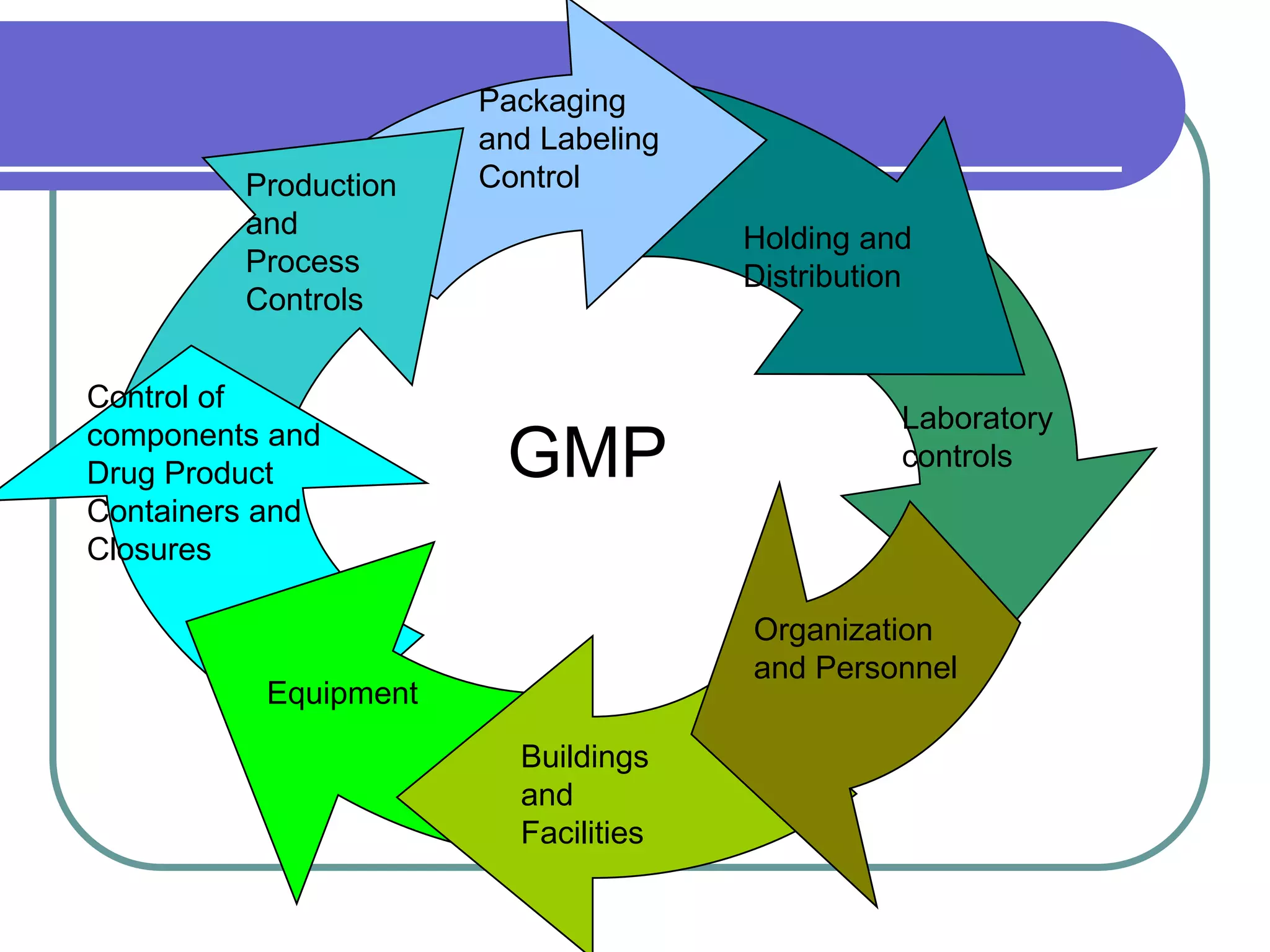










































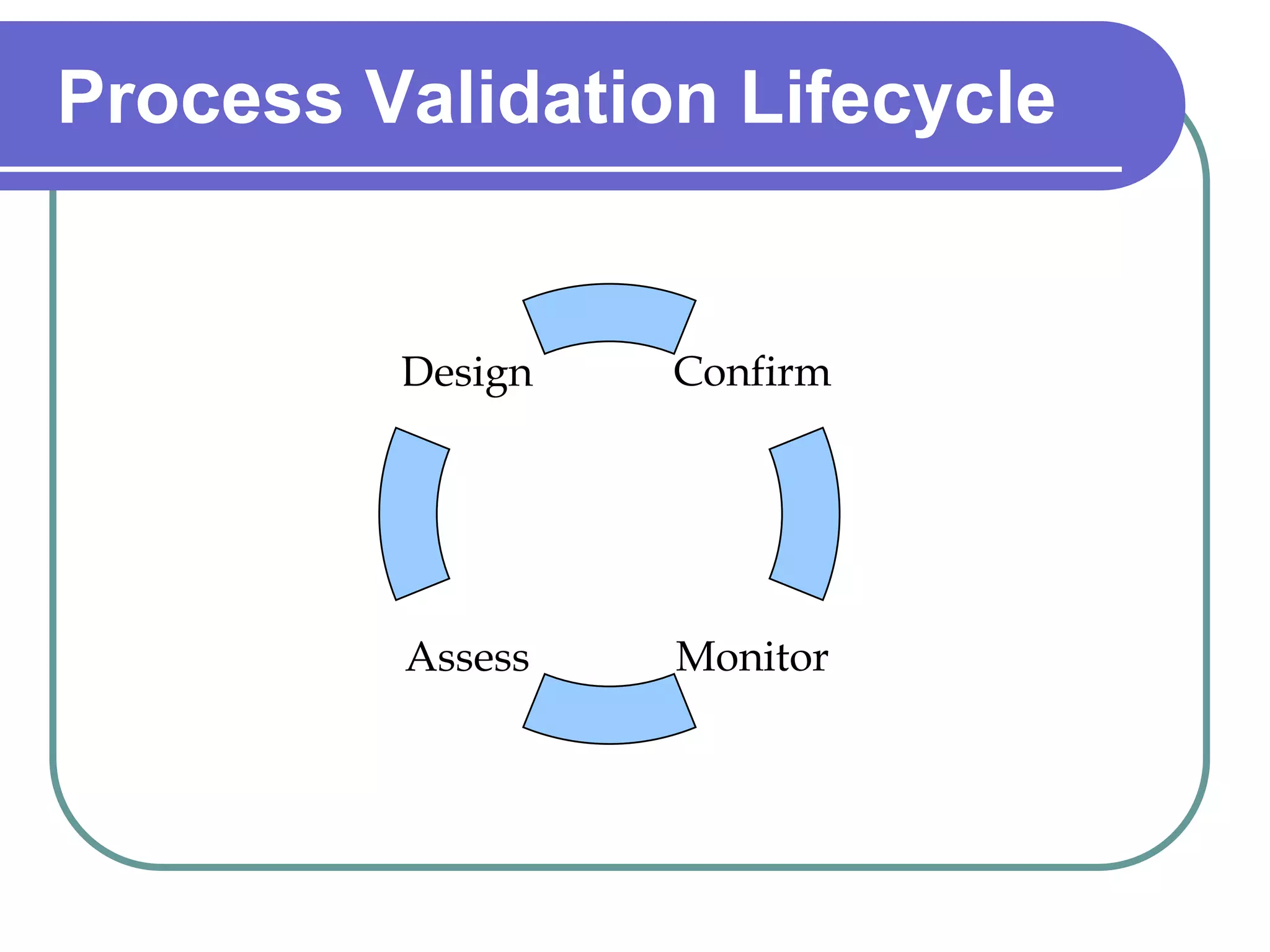





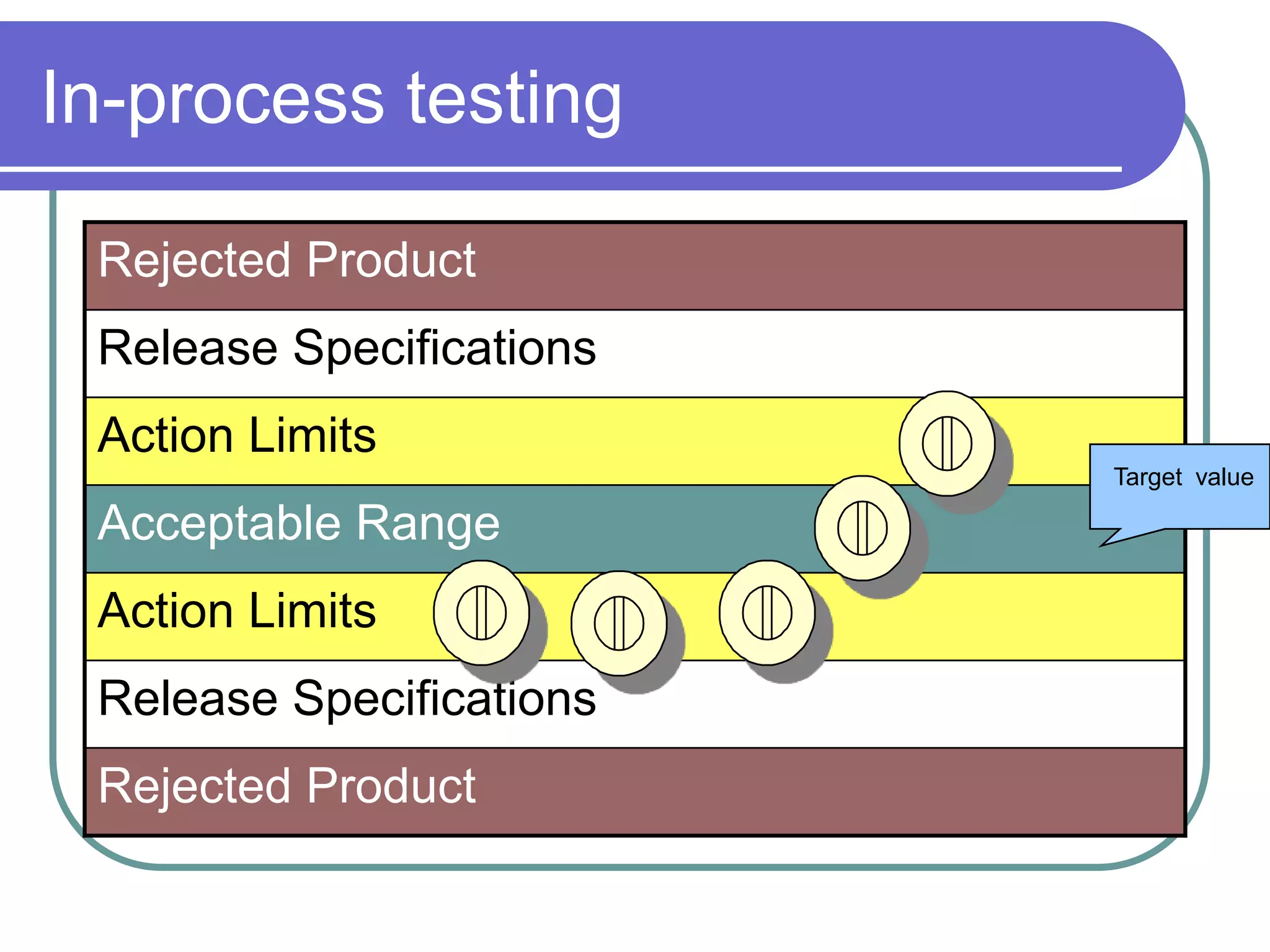






















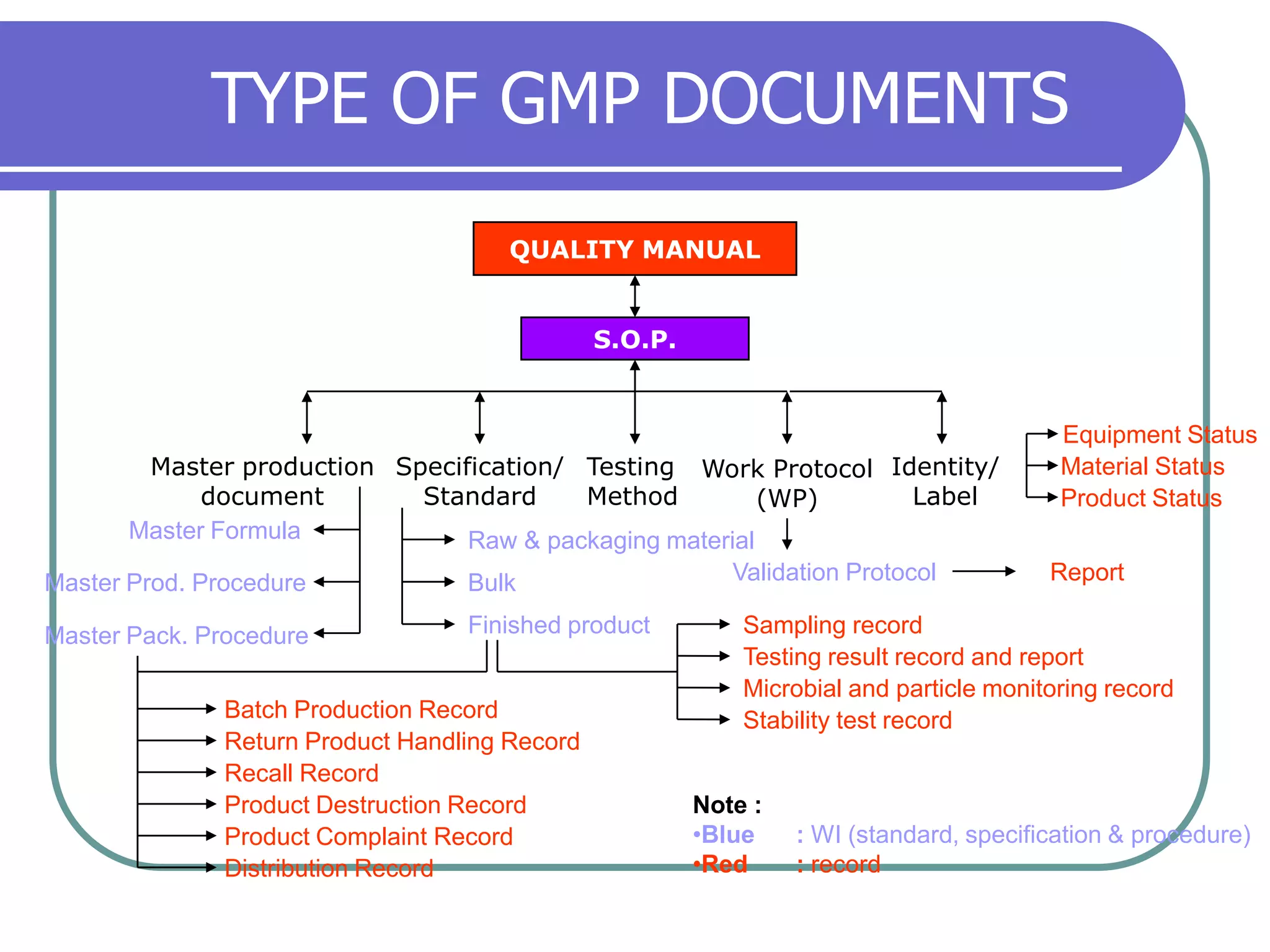

















The document discusses regulations governing Good Manufacturing Practices (GMP). It aims to provide an understanding of GMP regulations and their application and interpretation. The key GMP regulations cover the drug, medical device, food, and blood industries. GMP regulations provide flexibility for manufacturers but can also lead to confusion in interpretation. Subparts cover organization and personnel qualifications, facilities and equipment requirements, material and component controls, production and process controls, and documentation.

















































































