Download to read offline















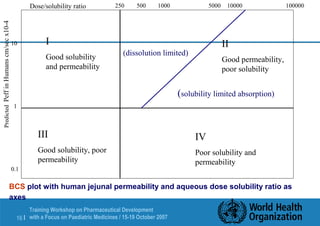





This document outlines an agenda for a training workshop on pharmaceutical development with a focus on paediatric formulations being held from October 15-19, 2007 in Tallinn, Estonia. The workshop will cover various topics including pre-formulation analytical studies, stress testing APIs, the impact of impurities on API specifications, excipient compatibility studies, degradation pathways, and the role of API processing in product instability. The goal is to discuss how preformulation studies influence key decisions in API and drug product development and support establishing appropriate container closure systems and analytical methods.













































































