Download to read offline








![Residual solvents
A) Class 1 (The most toxic and/or environmentally hazardous) [limited to 2–8
ppm]
Benzene Carbon tetrachloride 1,2-Dichloroethane 1,1-Dichloroethene 1,1,1-Trichloroethane
B) Class 2 (Considered a lesser risk) [ limit calculated by daily intake of theoretical
product mass of 10g]
Acetonitrile Chlorobenzene Chloroform Cumene Cyclohexane Cyclopentyl methyl ether 1,2-
Dichloroethene Dichloromethane 1,2-Dimethoxyethane N,N-Dimethylacetamide N,N-
Dimethylformamide 1,4-Dioxane 2-Ethoxyethanol Ethyleneglycol Formamide Hexane Methanol 2-
Methoxyethanol 0 Methylbutyl ketone Methylcyclohexane Methylisobutylketone3 N-Methylpyrrolidone
C) Class 3 (The lowest risk category)[ limited to 5000 ppm (0.5% w/w)]
1-Butanol Methyl acetate 1-Pentanol Ethanol Heptanes 1-Propanol Propyl acetate Acetone Isobutyl
acetate Tert-Butylmethyl ether Ethyl acetate 2-Butanol 3-Methyl-1- butanol Methyl isobutyl ketone Ethyl
ether Anisole Isopropyl acetate 2-Methyl-1-propanol 2-Propanol Acetic acid Ethyl format Cumene Methyl
ethyl ketone Pentane Formic acid Butyl acetate Dimethyl sulfoxide
8](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-8-320.jpg)
![9
Polymorphic forms
• Organic/inorganic
compounds form different
crystalline structures called
polymorphic forms. The
resulting change of
intermolecular interactions
gives rise to different
pharmacokinetic &
different properties of
organic and inorganic
materials
• In 2006 a new crystal form
of maleic acid had arisen
when solution of caffeine
and maleic acid (2:1) in
chloroform is set aside to
evaporate slowly
Genotoxic impurities (ICH M7
R1)
• Genotoxic compounds can
be carcinogenic to humans
due to their ability to
induce chromosomal
rearrangements and/or
genetic mutations.
• The ICH M7 (15) guideline
provides the limits for
control of genotoxic
impurities in
pharmaceuticals to limit
the risk of carcinogenicity.
Enantiomeric impurities
• To determine purity of the
chiral compound term
enantiomeric excess (EE) is
used.
• . These impurities present
in the drug are due to
change in the critical
parameter of molecules
during synthesis. The
following equation is used
to determine enantiomeric
excess (EE):
• EE= + [( R-S)/ ( R R)]×100
• where R and S stand for
the individual optical
isomer in the mixture
(and R+S=1)](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-9-320.jpg)















![Neotame
N-[N-(3,3-dimethylbutyl)-L-a-aspartyl]-L-phenylalanine .
It has extensive shelf life in dry conditions
7000-3000 sweeter than sucrose.
27](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-27-320.jpg)



![Procedure
Preparation of sample test solution:
1] Beverages
• Dilute 20 ml of the liquid in a 100 ml
volumetric flask with water.
• Filter the solution through a
membrane filter of pore size 0.2 um
before injection.
31](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-31-320.jpg)
![2]Juices, flavoured milk drinks:
32
• Dilute 20 ml sample with 50 ml water in a 100 ml volumetric flask.
• Add 2 ml Carrez solution 1, mix and 2 ml of Carrez solution 2, dilute to mark
with water and filter
• If the fat free insoluble matter in the initial sample mass exceeds approx 3 gm,
centrifuge the clarified solution for 10 minutes before filtering it quantitatively
into a 100 ml volumetric flask
• Wash the settled matter twice with water and centrifuge again
• Collect each of the supernatant in the 100 ml vol flask and then dilute the
solution with water](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-32-320.jpg)
![3] Jams and related products:
33
• Weigh to the nearest 1 mg, 20 gm of homogenized sample in a 100 ml volumetric flask
• Add about 60 ml water and place the flask in an ultrasonic bath at 40°C for 20 minutes
• The temperature should not exceed 40° C since aspartame can get degraded. Cool to room
temperature
• Add 2 ml Carrez solution I, mix followed by 2 ml carrez solution 2
• Add 2 ml Carrez solution I, mix followed by 2 ml carrez solution 2
• If the fat free insoluble matter in the initial mass exceeds 3 gm, is advisable to centrifuge the
clarified sample solution for 10 minutes at 1400 r.p.m it
• Wash with water and centrifuge again as in case of cloudy liquid samples](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-33-320.jpg)
![4] Semisolid and solid products:
34
• Weigh 10-20 gm of thoroughly homogenized sample in a 100
ml volumetric flask.
• Add about 50 ml water and place the vol flask in an ultra sonic
bath at 40°C for 20 minutes
• Cool to room temperature, add 2 ml Carrez solution 1, mix and
add 2 ml of Carrez solution 2, dilute with water and filter
• In case of very complex matrices, additional purification using
the solid phase extraction column
• In this case add 2 ml of clarified filterate to the cartridge,
previously activated with 3 ml of methanol and 20 ml water and
elute with about 20 ml of mobile phase](https://image.slidesharecdn.com/impurityandfoodagents-230730070738-9c66054f/85/Impurity-and-food-agents-seminar-pptx-34-320.jpg)









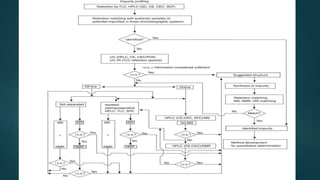
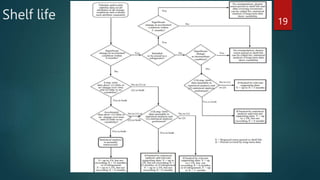
The document discusses the importance of impurity profiling and degradation characterization in pharmaceuticals, highlighting the regulatory scrutiny that necessitates these analyses due to potential impacts on drug quality and safety. It covers various types of impurities, methods for identification, and the crucial role of stability testing in determining shelf life and re-test periods for drugs. Additionally, it includes the analysis of artificial sweeteners and their regulatory status, profiling techniques, and specific methodologies for identifying and quantifying these substances.





















![Basics impurity profiling and degradent characterization[134]](https://cdn.slidesharecdn.com/ss_thumbnails/basicsimpurityprofilinganddegradentcharacterization134-191014164210-thumbnail.jpg?width=640&height=640&fit=bounds)



































