Download to read offline
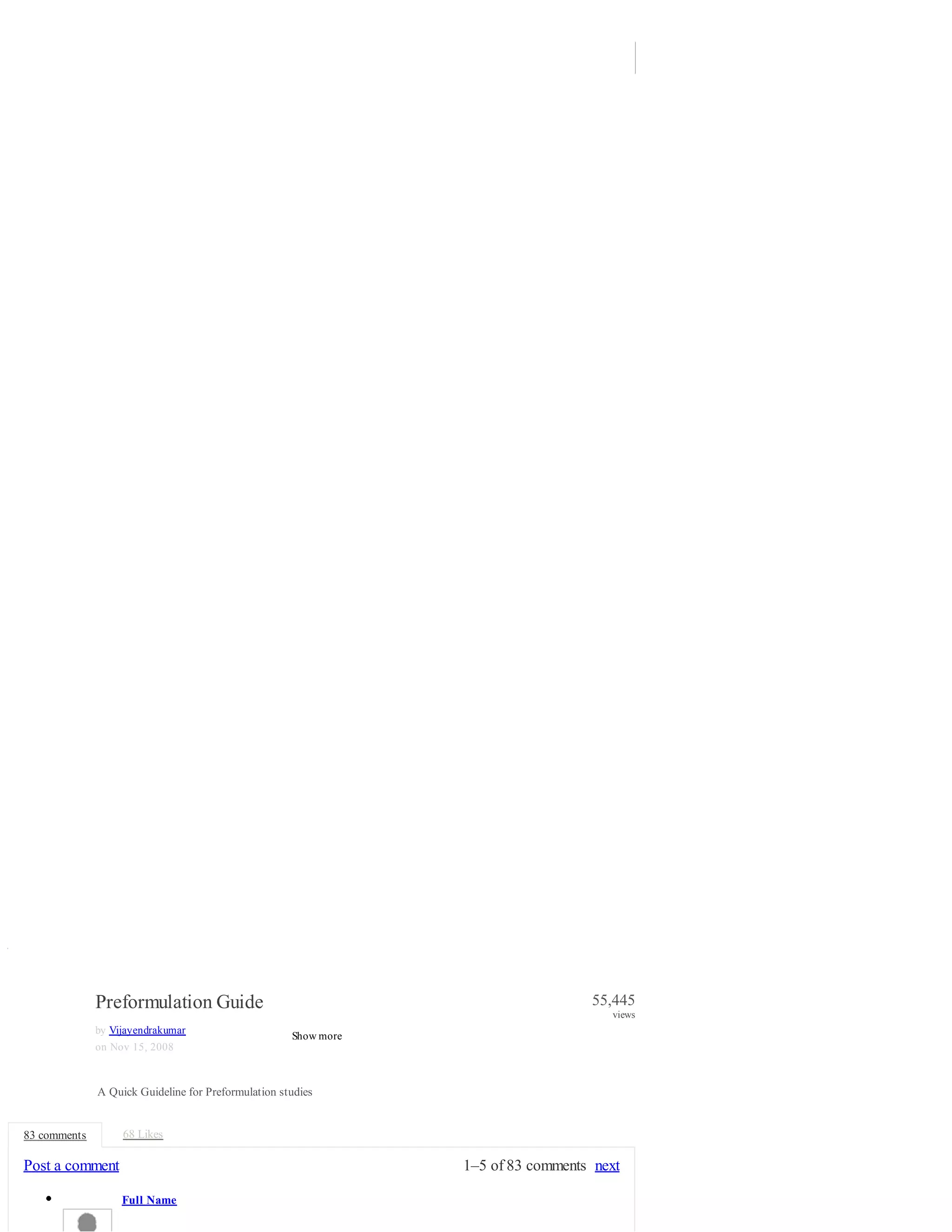
![Preclinical Testing Involves: 1. Pharmacology 2. Toxicology 3. Preformulation 4. Formulation 5.
Analytical
3. PREFORMULATION GUIDANCE 6. Pharmacokinetics 1. Pharmacology: 2. Toxicology: Toxicology
studies in preclinical stage are conducted to: • Select or reject lead candidate • General Indication of
suitability • Dose selection and guidance to clinician The basic studies conducted are: • Mutagenicity - In
vitro Ames test [ A test used to determine the mutagenic potential of a substance based on the mutation
rate of bacteria that are exposed to the substance] . • One-week or Two-week Range Finders: Mouse
or Rat, Dog or Primate • Maximum Tolerated Dose • Gross Effects, Clinical Chemistries - What are the
toxicities • Gross Pathology to Indicate Target Organs • Satisfactory Therapeutic Ratio vis-a-vis Animal
Efficacy Studies Single Dose Acute Toxicity Testing for Pharmaceuticals (Refer CDER Guidelines) 3.
Preformulation : Preformulation involves the application of biopharmaceutical principles to the
physicochemical parameters of a drug with the goal of designing an optimum drug delivery system.
Characterization of drug molecule is a very important step at the preformulation phase of product
development. Following studies are conducted
4. PREFORMULATION GUIDANCE as basic preformulation studies; special studies are conducted
depending on the type of dosage form and the type of drug molecule: 1. Solubility determination 2. pKa
determination 3. Partition coefficient 4. Chemical stability profile 5. Crystal Properties and Polymorphism
6. Particle size, shape and surface area 7. Specifications for New Drug Substances and Products 8.
Dosage Form Development Chart 1. Solubility determination The solubility of drug is an important
physicochemical property because it affects the bioavailability of the drug, the rate of drug release into
the dissolution medium, and consequently, the therapeutic efficacy of the pharmaceutical product. The
solubility of a molecule in various solvents is determined as a first step. This information is valuable in
developing a formulation. Solubility is usually determined in a variety of commonly used solvents and
some oils if the molecule is lipophilic. The solubility of a material is usually determined by the equilibrium
solubility method, which employs a saturated solution of the material, obtained by stirring an excess of
material in the solvent for a prolonged period until equilibrium is achieved.
5. PREFORMULATION GUIDANCE Common solvents used for solubility determination are: I. Water II.
Polyethylene Glycols III. Propylene Glycol IV. Glycerin V. Sorbitol VI. Ethyl Alcohol VII. Methanol VIII.
Benzyl Alcohol IX. Isopropyl Alcohol X. Tweens XI. Polysorbates XII. Castor Oil XIII. Peanut Oil XIV.
Sesame Oil XV. Buffers at various pHs 2. pKa determination Determination of the dissociation constant
for a drug capable of ionization within a pH range of 1 to 10 is important since solubility, and
consequently absorption, can be altered by orders of magnitude with changing pH. The Henderson-
Hasselbalch equation provides an estimate of the ionized and un-ionized drug concentration at a particular
pH. For acidic compounds: pH = pKa + log ([ionized drug]/[un-ionized drug]) For basic compounds: pH
= pKa + log ([un-ionized drug]/[ionized drug])
6. PREFORMULATION GUIDANCE pKa of a compound is thus a measure of drug un-ionized at a
certain pH pKa = -log K a , where Ka is the acidity or ionization constant of a weak acid. (K w=[H 3 O+]
For a weak base, K a = K w/K b , where K w is the ionic product of water x [OH-]) and K b is the
basicity or ionization constant of the weak 3. Partition coefficient Partition coefficient (oil/water) is a
measure of a drug's lipophilicity and an indication of its ability to cross cell membranes. It is defined as
the ratio of un- ionized drug distributed between the organic and aqueous phases at equilibrium. P o/w =
(C oil /C water ) equilibrium For series of compounds, the partition coefficient can provide an empiric
handle in screening for some biologic properties. For drug delivery, the lipophilic/hydrophilic balance has
been shown to be a contributing factor for the rate and extent of drug absorption. Although partition
coefficient data alone does not provide understanding of in vivo absorption, it does provide a means of
characterizing the lipophilic/hydrophilic nature of the drug. Since biological membranes are lipoidal in
nature, the rate of drug transfer for passively absorbed drugs is directly related to the lipophilicity of the
moleucle. The partition coefficient is commonly determined using an oil phase of octanol or chloroform
and water. Drugs having values of P much greater than 1 are classified as lipophilic, whereas those with
partition coefficients much less than 1 are indicative of a hydrophilic drug. Although it appears that the
partition coefficient may be the best predictor of absorption rate, the effect of dissolution rate, pKa, and
solubility on absorption must not be neglected.
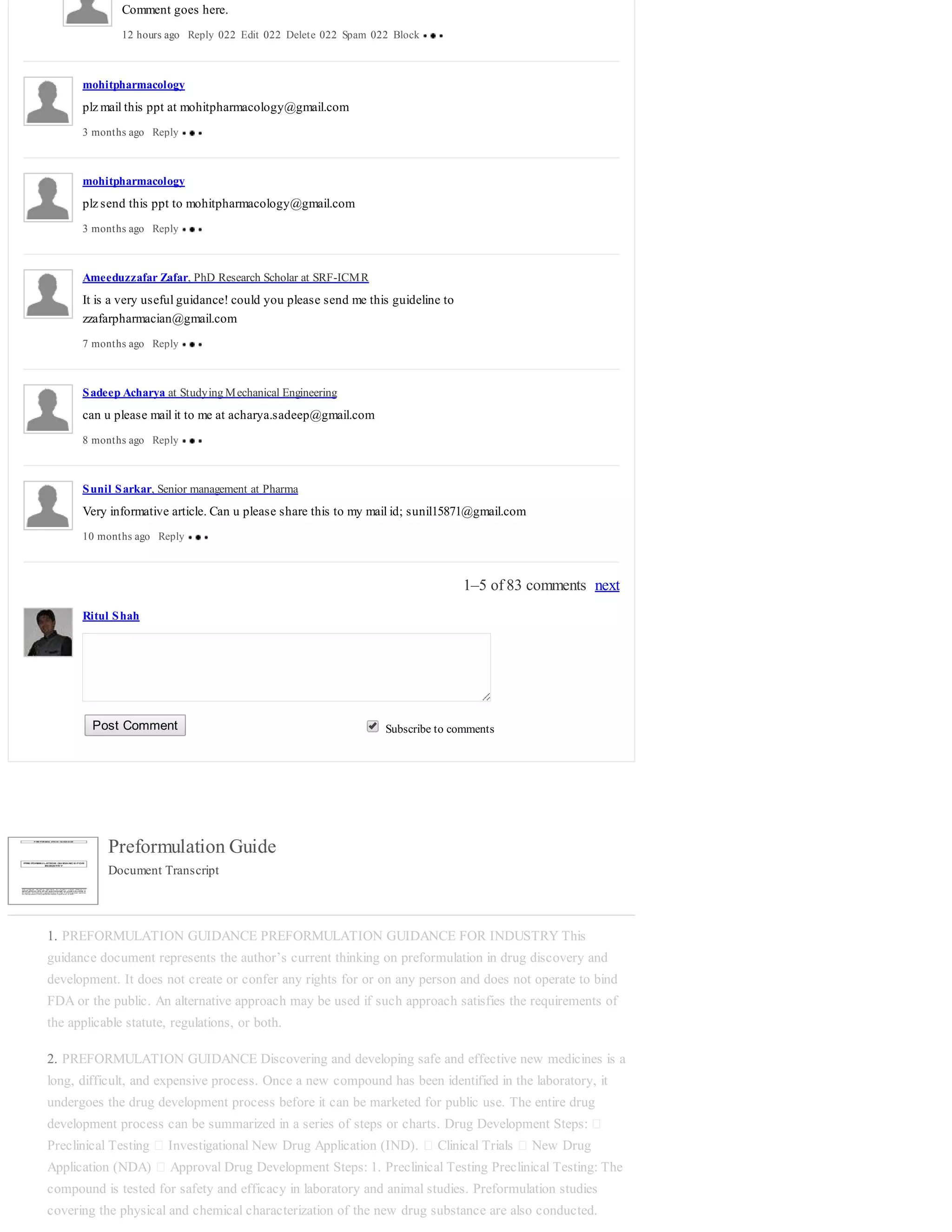
7. PREFORMULATION GUIDANCE 4. Chemical stability profile Preformulation stability studies are](https://image.slidesharecdn.com/preformulationguide-140702091334-phpapp02/75/Preformulation-guide-3-2048.jpg)


![mixture both quot;as isquot; and granulated with water at elevated temperatures. The granulation may be
carried out so that the mixture contains fixed amounts (e.g., 5%) of moisture. The mixtures are sealed in
ampoules to prevent any escape of moisture at elevated temperatures. The samples are examined
periodically for appearance and analyzed for any decomposition using thin-layer chromatography.
Unstressed samples are used as controls. Any change in the chromatograph such as the appearance of a
new spot
15. PREFORMULATION GUIDANCE or a change in the RF values [The distance (d s ) traveled by a
sample, relative to the solvent front (d e ) which is compound specific in each chromatographic system.
The ratio is termed the R f value or resolution factor. This is sometimes known as relative to front.] of
the components is indicative of an interaction. The technique may be quantities if deemed necessary. If
significant interaction is noticed at elevated temperatures, corroborative evidence must be obtained by
examining mixtures stored at lower temperatures for longer durations. If no interaction is observed at 60
or 70°C, especially in the presence of moisture, none can be expected at lower temperatures. Among the
advantages of thin-layer chromatographies in this application are: • Evidence of degradation is
unequivocal. • The spots corresponding to degradation products can be eluted for possible identification.
The technique can be quantitated to obtain kinetic data. • Differential Thermal Analysis: Thermal Analysis
is useful in the investigation of solid-state interactions. It is also useful in the detection of eutectics.
Thermograms are generated for pure components and their physical mixtures with other components. In
the absence of any interaction, the thermograms of mixtures show patterns corresponding to those of the
individual components. In the event that interaction occurs, this is indicated in the thermogram of a
mixture by the appearance of one or more new peaks or the disappearance of one or more peaks
corresponding to those of the components.
16. PREFORMULATION GUIDANCE Diffuse Reflectance Spectroscopy: Diffuse reflectance
spectroscopy is gaining increasing popularity among preformulation scientists as a tool to detect and
monitor drug-excipient interactions. In this technique solid drug, excipients, and their physical mixtures
are exposed to incident radiation. A portion of the incident radiation is partly absorbed and partly reflected
in a diffuse manner. The diffuse reflectance depends on the packing density of the solid, its particle size,
and its crystal form, among other factors. When these factors are adequately controlled, diffuse
reflectance spectroscopy can be used to investigate physical and chemical changes occurring on solid
surfaces. A shift in the diffuse reflectance spectrum of the drug due to the presence of the excipient
indicates physical adsorption, whereas the appearance of a new peak indicates chemisorption or
formation of a degradation product.
17. PREFORMULATION GUIDANCE Typical Stability Protocol for a New Chemical Entity
18. PREFORMULATION GUIDANCE
19. PREFORMULATION GUIDANCE 5. Crystal Properties and Polymorphism Many drug substances
can exist in more than one crystalline form with different space lattice arrangements. This property is
known as polymorphism. Polymorphs generally have different melting points, x-ray diffraction patterns,
and solubilities, even though they are chemically identical. Differences in the dissolution rates and
solubilities of different polymorphic forms of a given drug are very commonly observed. When the
absorption of a drug is dissolution rate limited, a more soluble and faster-dissolving form may be utilized
to improve the rate and extent of bioavailability. For drugs prone to degradation in the solid state, the
physical form of the drug influences degradation. Selection of a polymorph that is chemically more stable
is a solution in many cases. Different polymorphs also lead to different morphology, tensile strength and
density of powder bed which all contribute to compression characteristics of materials. Some
investigation of polymorphism and crystal habit of a drug substance as it relates to pharmaceutical
processing is desirable during its preformulation evaluation, especially when the active ingredient is
expected to constitute the bulk of the tablet mass. Although a drug substance may exist in two or more
polymorphic forms, only one form is thermodynamically stable at a given temperature and pressure. The
other forms would convert to the stable form with time. In general, the stable polymorph exhibits the
highest melting point, the lowest solubility, and the maximum chemical stability. Various techniques are
available for the investigation of the solid state. These include microscopy (including hot-stage
microscopy), infrared spectrophotometry, single-crystal X-ray and X-ray powder diffraction, thermal
analysis, and dilatometry.](https://image.slidesharecdn.com/preformulationguide-140702091334-phpapp02/75/Preformulation-guide-6-2048.jpg)





1. The document provides guidance on preformulation studies for new drug candidates. Preformulation involves characterizing the physical and chemical properties of a drug substance to aid in developing an optimal dosage form. 2. Key preformulation studies discussed include determining solubility, pKa, partition coefficient, chemical stability, and polymorphism. These studies provide important information on factors like bioavailability and dosage form design. 3. The guidance describes common techniques for conducting preformulation studies like equilibrium solubility methods and stresses the importance of stability testing under various conditions like temperature, pH and light exposure.






















































































