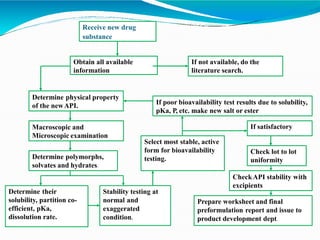
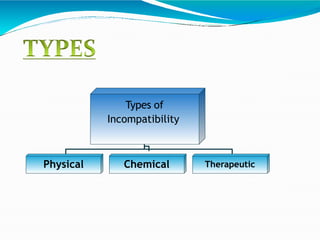
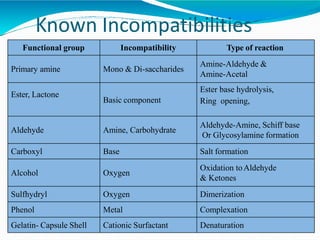
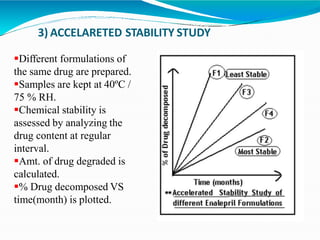
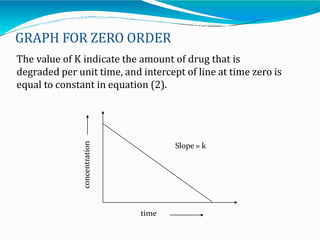
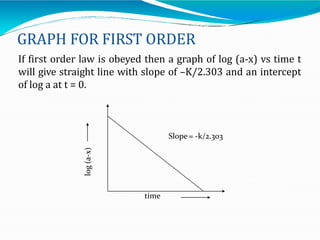
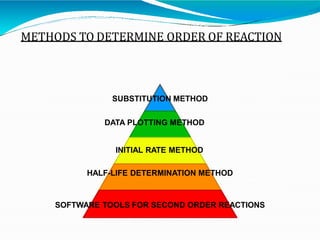
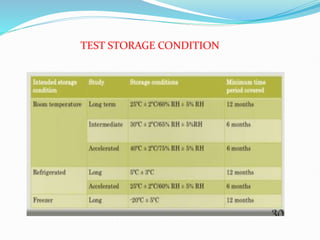
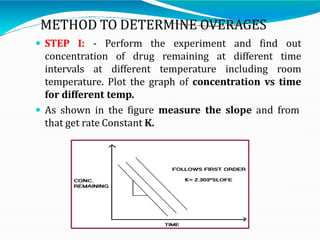
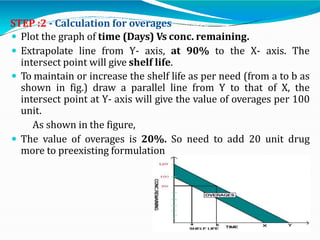
Preformulation is defined as the investigation of physical and chemical properties of a drug substance alone and when combined with excipients. The goal is to generate information to help formulators develop stable and safe dosage forms with good bioavailability. Some key tests include determining the drug's solubility, stability, and compatibility with various excipients using techniques like DSC, TLC, and HPLC. This provides critical data to guide the rational selection of dosage form and formulation components.