Downloaded 342 times





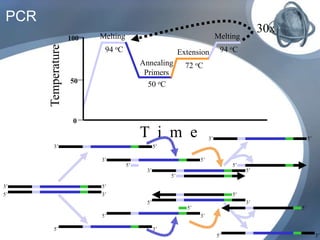
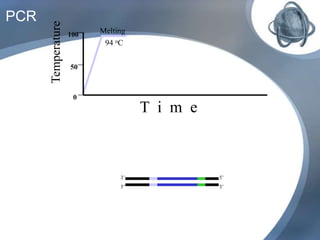
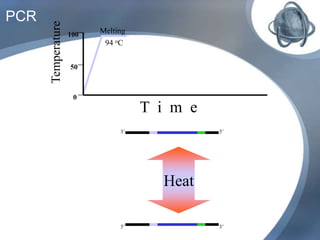

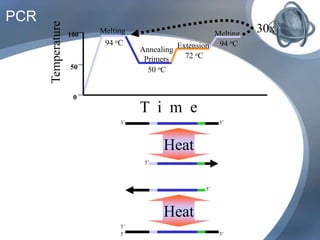


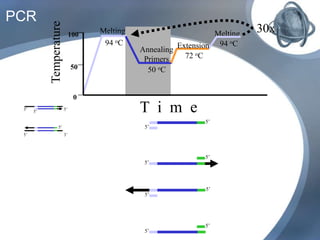
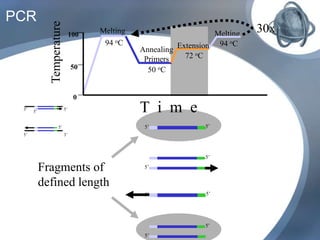
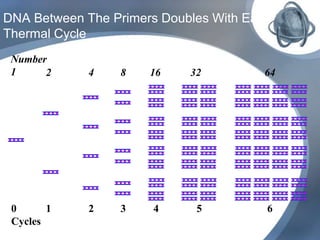













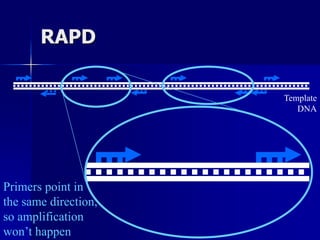
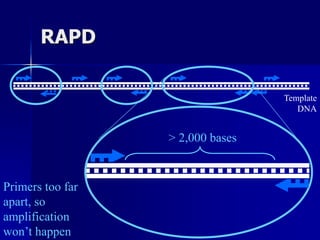
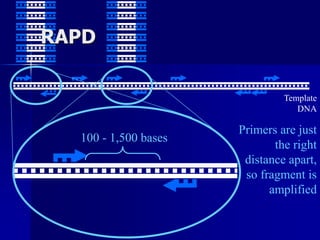
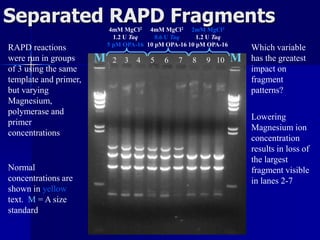





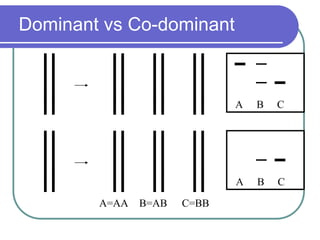

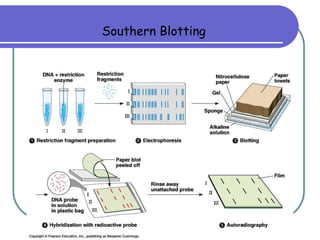
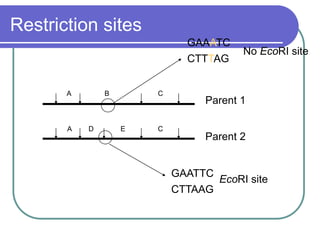
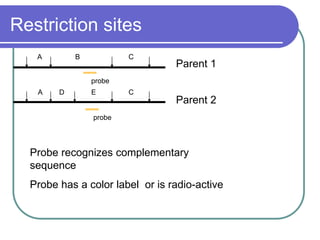
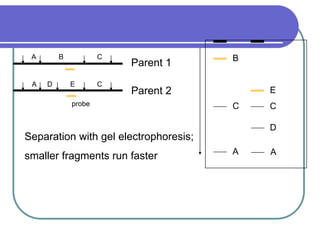
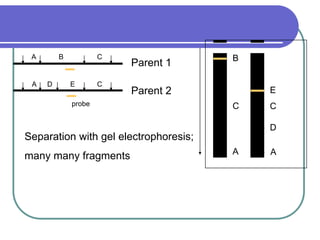
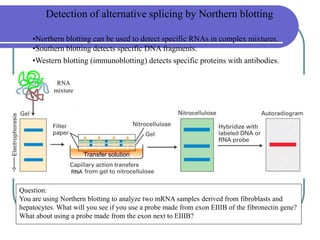

The document outlines the principles and processes of three genetic techniques: Polymerase Chain Reaction (PCR), Random Amplified Polymorphic DNA (RAPD), and Restriction Fragment Length Polymorphism (RFLP). It provides detailed information on PCR methodology, including temperature cycles and primer design, as well as how RAPD uses short primers to generate genetic markers. Additionally, it discusses RFLP's reliance on restriction enzymes for detecting genetic variations through electrophoresis and hybridization techniques.










































































![[DSC Europe 25] Andrzej Kowalczyk - AI - how to start small and grow in the f...](https://cdn.slidesharecdn.com/ss_thumbnails/oy1zmo94qv6vpcqjvno2-andrzej-kowalczyk-ai-how-to-start-small-and-grow-in-the-future-1-260119121559-cf093b23-thumbnail.jpg?width=640&height=640&fit=bounds)





![[DSC Europe 25] Bojan Djuricic - Predictive Design Process.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/5awdrbedqdek3gqu2ezy-4-the-predictive-design-bojan-djuricic-260120105856-6c399e9b-thumbnail.jpg?width=640&height=640&fit=bounds)






