Downloaded 128 times








































![Knowledge-based Scoring
Function
Free energies of molecular interactions
derived from structural information on
Protein-ligand complexes contained in PDB
( ) ( )[ ]lpreflp FPP σσβσσ ,exp, −=
Boltzmann-Like Statistics of Interatomic
Contacts.](https://image.slidesharecdn.com/moleculardocking-171012130519/85/Molecular-docking-42-320.jpg)



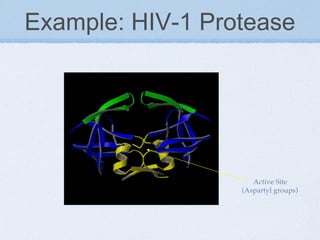
Molecular docking involves determining the optimal orientation of two biological molecules to maximize their interaction and minimize their total energy. It is important for understanding protein-protein interactions and rational drug design. Docking programs represent molecule surfaces and match them to find orientations, evaluating via scoring functions like shape complementarity, empirical potentials, or knowledge-based potentials derived from protein structures. Common techniques include representing surfaces as spheres or alpha shapes, and optimization methods like Monte Carlo, molecular dynamics, or genetic algorithms to introduce flexibility.























































































