
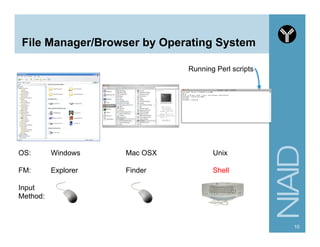
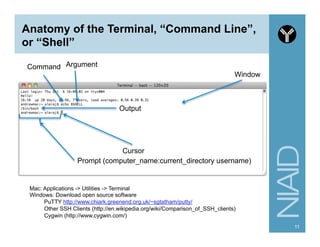
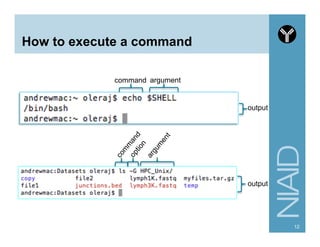
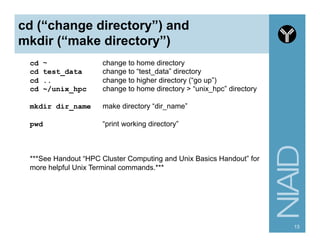
Download as PDF, PPTX













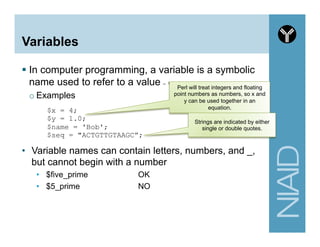



![Common Scalar Functions Examples
my $string = "This string has a newline.n";
chomp $string;
print $string;
#prints "This string has a newline.”
$string = lc($string);
print $string;
#prints ”this string has a newline.”
@array = split(" ", $string);
#array looks like [“this", "string", "has",
"a", "newline."]](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-22-320.jpg)

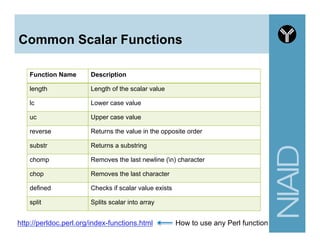
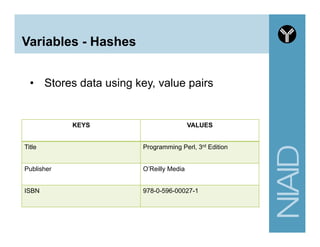
![Variables - Array
• Begins with @
• Use the () brackets for creating
• Use the $ and [] brackets for retrieving a single
element in the array
my @grades = (75, 80, 35);
my @mixnmatch = (5, "A", 4.5);
my @names = ("Bob", "Vivek", "Jane");
# zero-based index
my $first_name = $names[0];
# retrieve the last item in an array
my $last_name = $names[-1];](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-25-320.jpg)





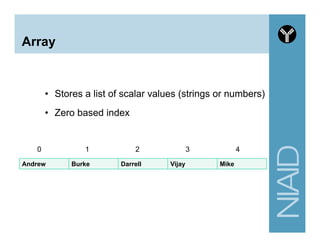
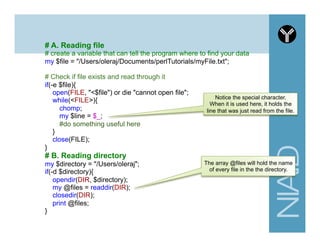
![Variables summary
# A. Scalar variable
my $first_name = "andrew";
my $last_name = "oler”;
# B. Array variable
# use 'circular' bracket and @ symbol for assignment
my @personal_info = ("andrew", $last_name);
# use 'square' bracket and the integer index to access an entry
my $fname = $personal_info[0];
# C. Hash variable
# use 'circular' brackets (similar to array) and % symbol for assignment
my %personal_info = (
first_name => "andrew",
last_name => "oler"
);
# use 'curly' brackets to access a single entry
my $fname1 = $personal_info{first_name};](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-33-320.jpg)














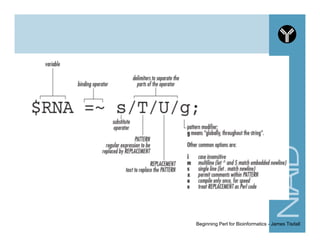

![Regular Expressions (REGEX)
Symbol Meaning
. Match any one character (except
newline).
^ Match at beginning of string
$ Match at end of string
n Match the newline
t Match a tab
s Match any whitespace character
w Match any word
character (alphanumeric plus "_")
W Match any non-word character
d Match any digit character
[A-Za-z] Match any letter
[0-9] same as d
my $string = "See also xyz";
$string =~ /See also ./;
#matches "See also x”
$string =~ /^./;
#matches "S”
$string =~ /.$/;
#matches "z”
$string =~ /wsw/;
#matches "e a"
http://www.troubleshooters.com/codecorn/littperl/perlreg.htm](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-51-320.jpg)

![REGEX Examples
my $string = ">ref|XP_001882498.1| retrovirus-related pol polyprotein
[Laccaria bicolor S238N-H82]";
$string =~/s.*virus/;
#will match " retrovirus"
$string =~ /XP_d+/;
#will match "XP_001882498”
$string =~ /XP_d/;
#match “XP_0”
$string =~ /[.*]$/;
#will match "[Laccaria bicolor S238N-H82]"
$string =~ /^.*|/;
#will match ">ref|XP_001882498.1|"
$string =~ /^.*?|/;
#will match ">ref|"
$string =~ s/|/:/g;
#string becomes ">ref:XP_001882498.1: retrovirus-related pol polyprotein
[Laccaria bicolor S238N-H82]"](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-53-320.jpg)







![Creating a Module
#!/usr/bin/perl!
!
package Foo;!
sub bar {!
print "Hello $_[0]n"!
}!
!
sub blat {!
print "World $_[0]n"!
}!
1;
61](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-61-320.jpg)


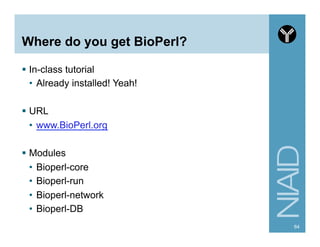


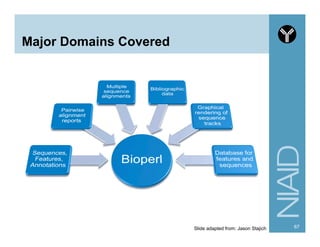
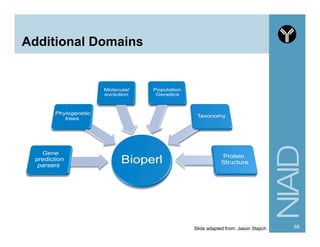




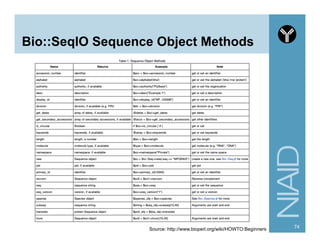





![Module:
Bio::SearchIO Methods
80http://www.bioperl.org/wiki/HOWTO:SearchIO
Method
Example
Description
algorithm
BLASTX
algorithm string
algorithm_version
2.2.4 [Aug-26-2002]
algorithm version
query_name
20521485|dbj|AP004641.2
query name
query_accession
AP004641.2
query accession
query_length
3059
query length
query_description
Oryza sativa ... 977CE9AF checksum.
query description
database_name
test.fa
database name
database_letters
1291
number of residues in database
database_entries
5
number of database entries
available_statistics
effectivespaceused ... dbletters
statistics used
available_parameters
gapext matrix allowgaps gapopen
parameters used
num_hits
1
number of hits](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-80-320.jpg)



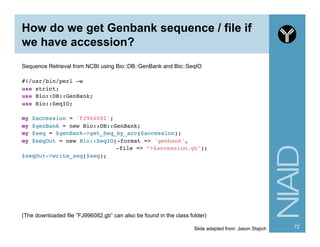
![Finding Motifs in Sequences
#!/usr/bin/perl -w!
use strict;!
use Bio::SeqIO;!
!
my $file = 'hsp.fas';!
my $motif = "[ATG]A";!
#my $motif = '(A[^T]{2,}){2,}’;!
!
my $in = Bio::SeqIO->new(-format => 'fasta', -file => $file);!
my $motif_count = 0;!
!
while ( my $seq = $in->next_seq) {!
!my $str = $seq->seq; ! !# get the sequence as a string!
!if ( $str =~ /$motif/i ) {!
! !$motif_count++; # of sequences that have this motif!
!}!
}!
!
printf "%d sequences have the motif $motifn", $motif_count;
84](https://image.slidesharecdn.com/2014introperlandbioperlniaid-170502191439/85/Introduction-to-Perl-and-BioPerl-84-320.jpg)





This document provides an overview of using the Perl programming language for bioinformatics applications. It introduces Perl variables like scalars, arrays, and hashes. It also covers flow control and loops. The document demonstrates how to open and read/write files in Perl. It provides examples of commonly used bioinformatics tools that incorporate Perl components and recommends resources for learning more about Perl and BioPerl.























































































