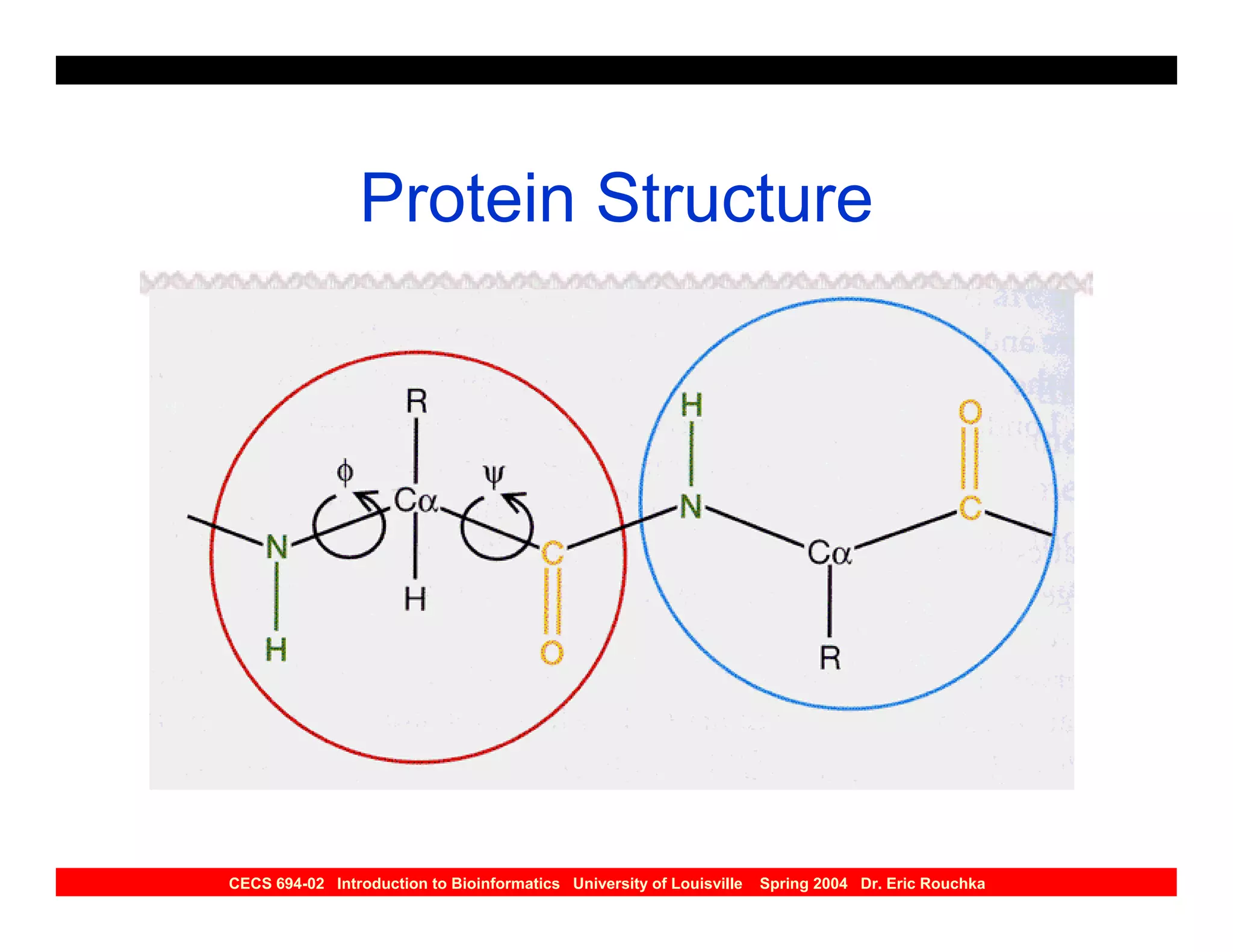
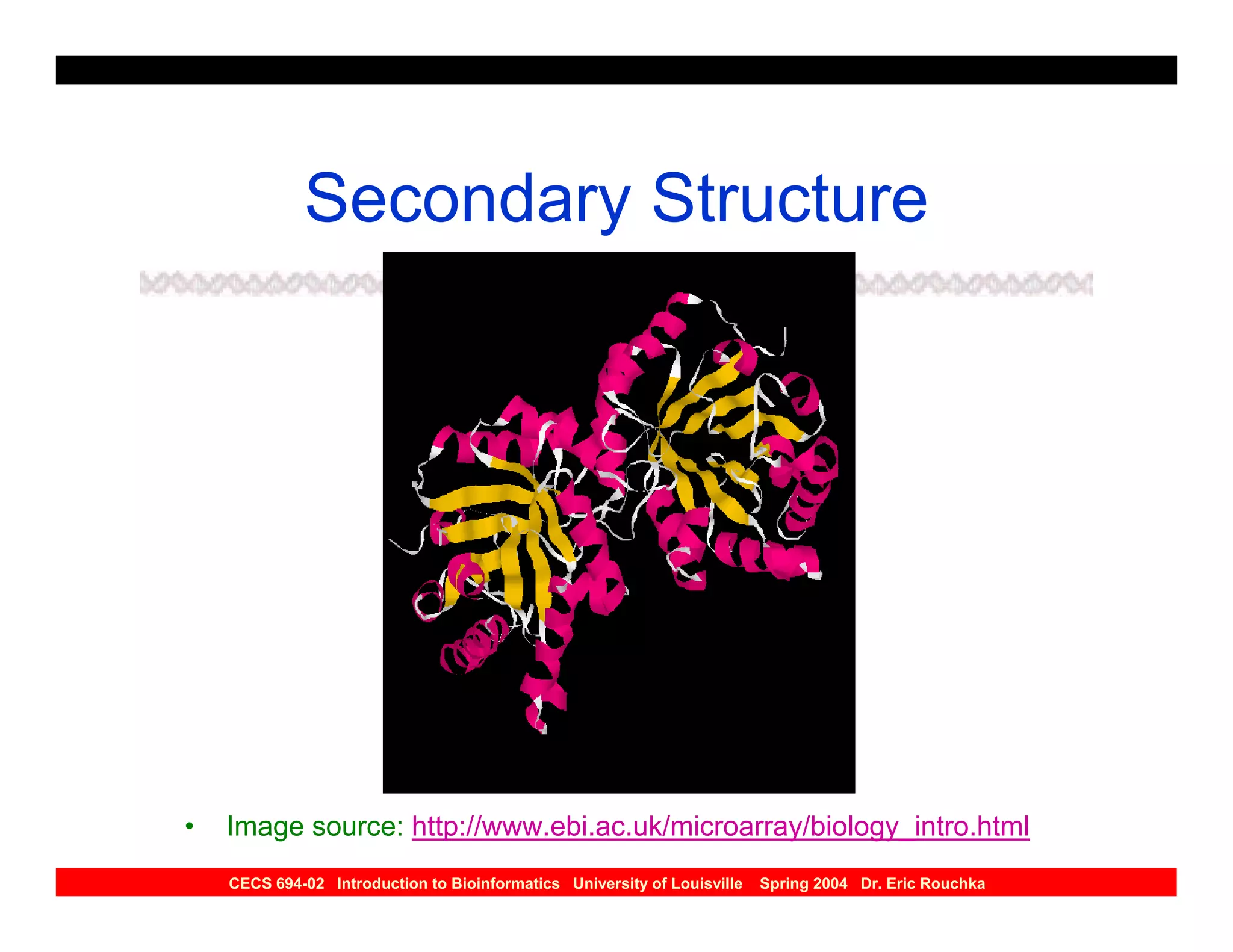
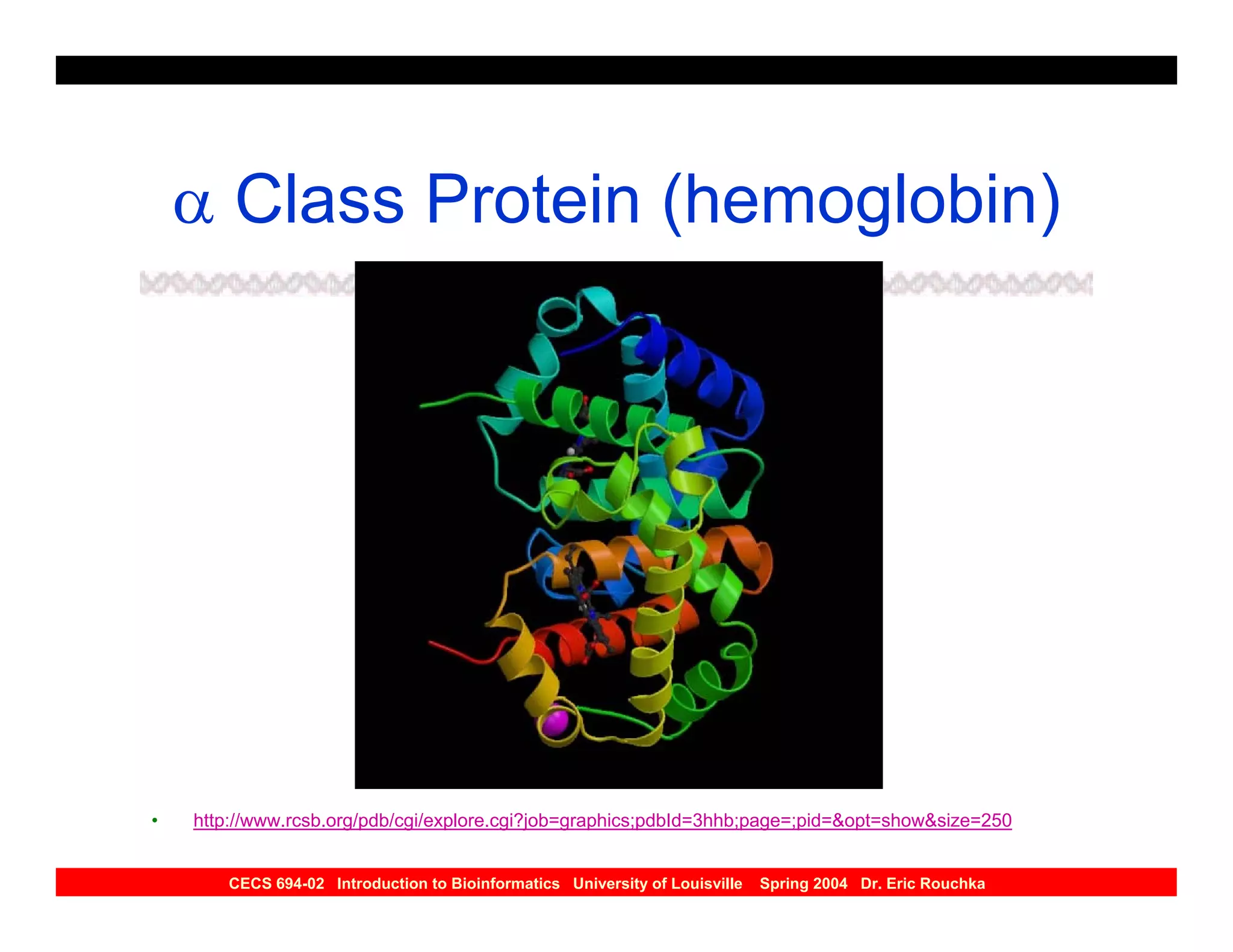
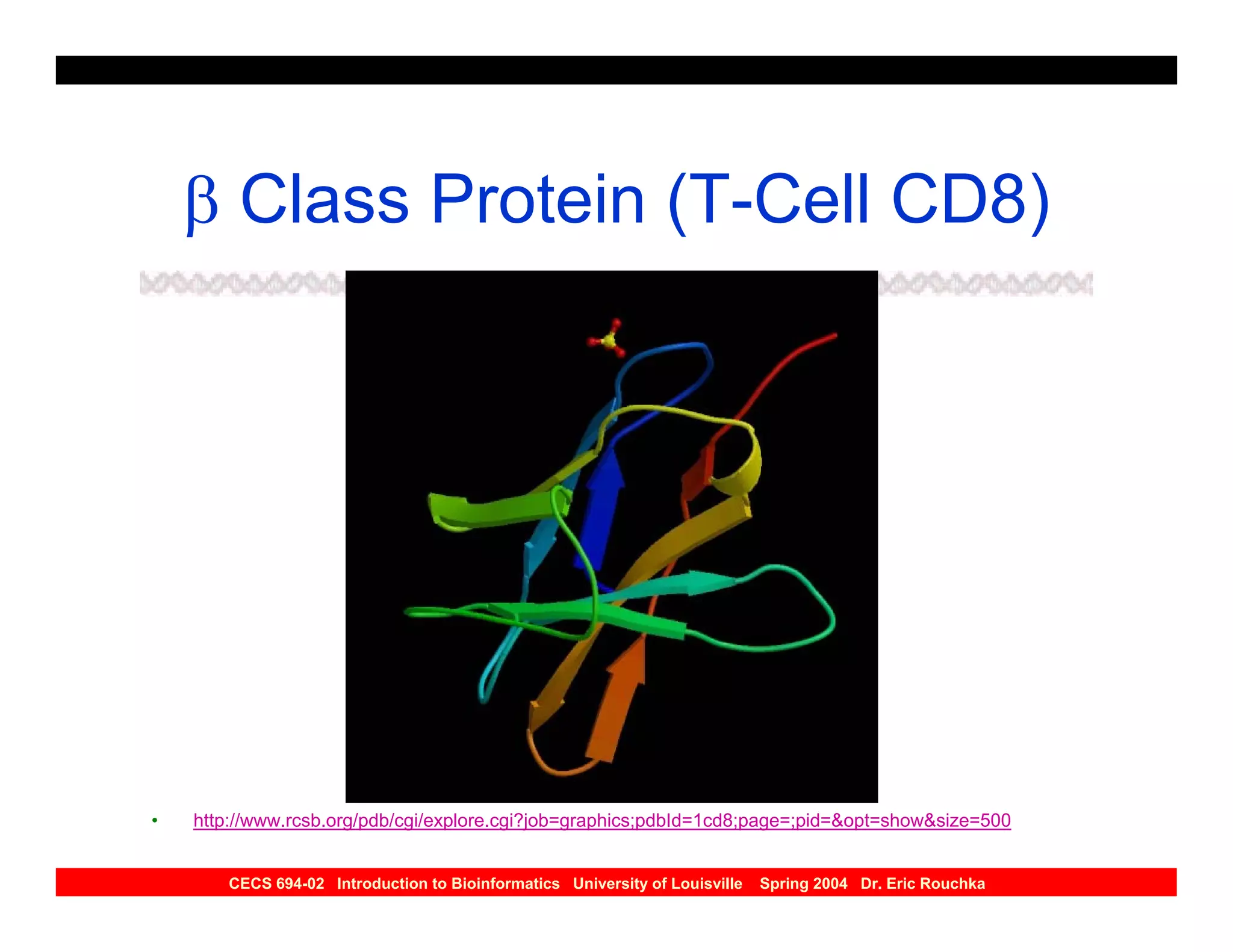
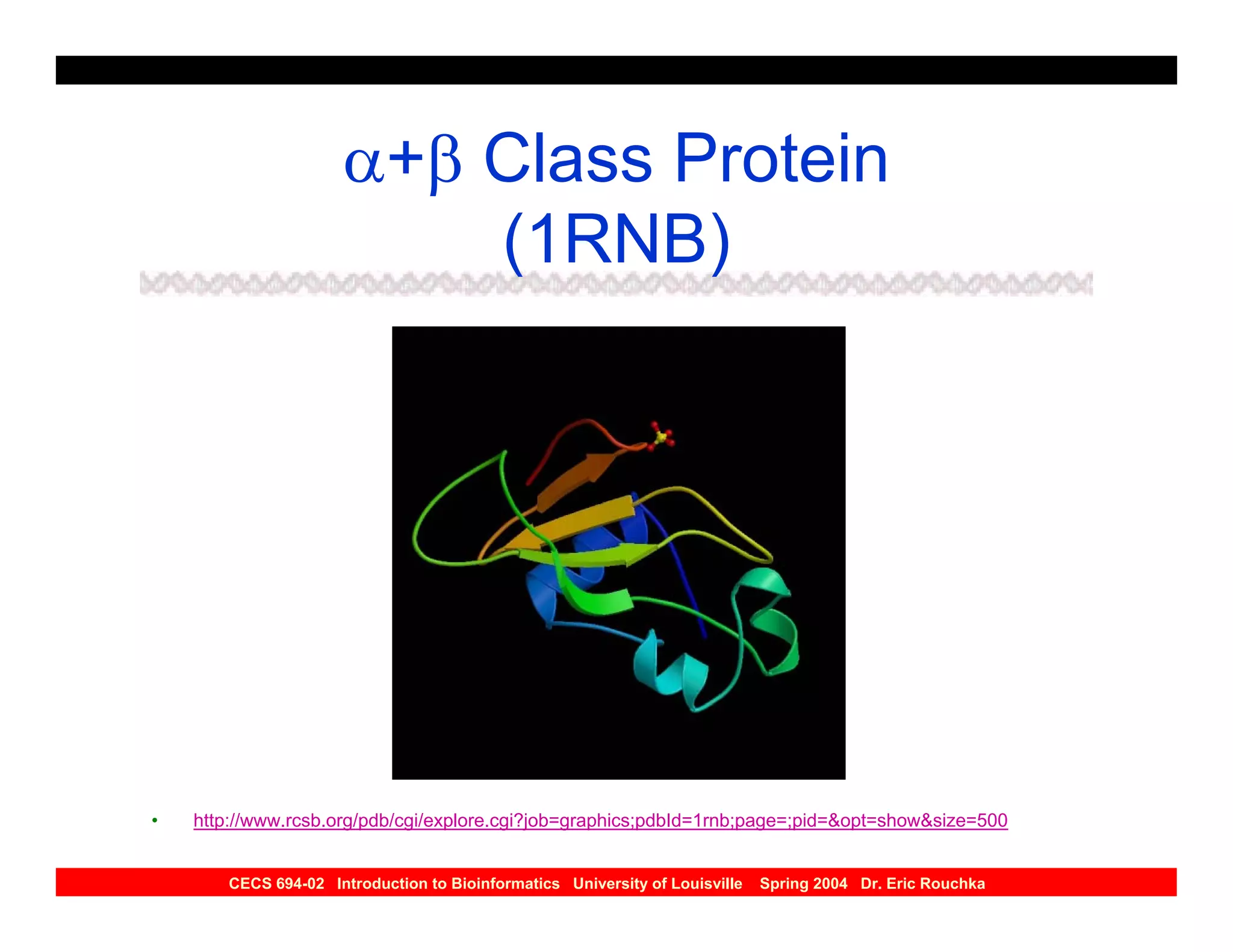
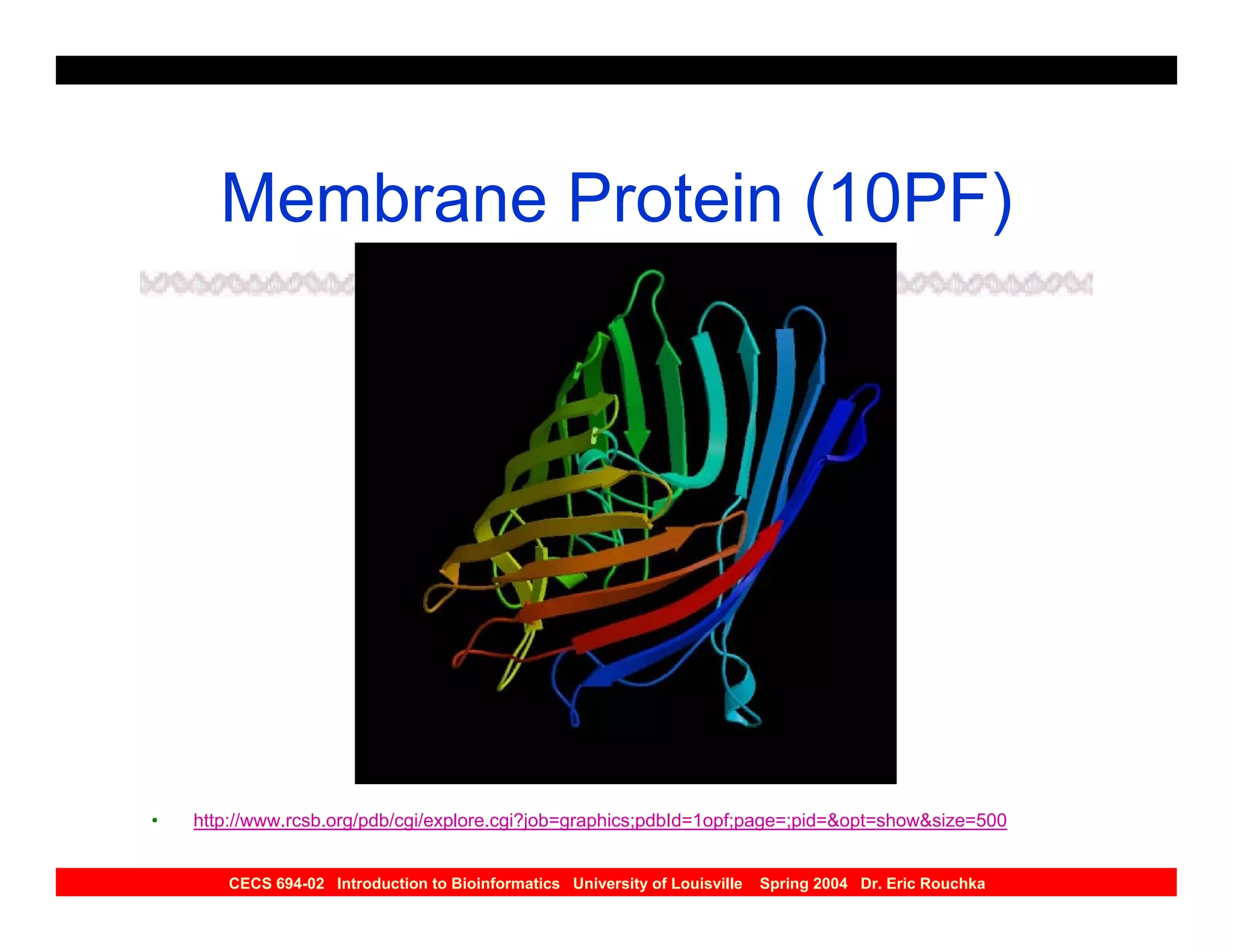
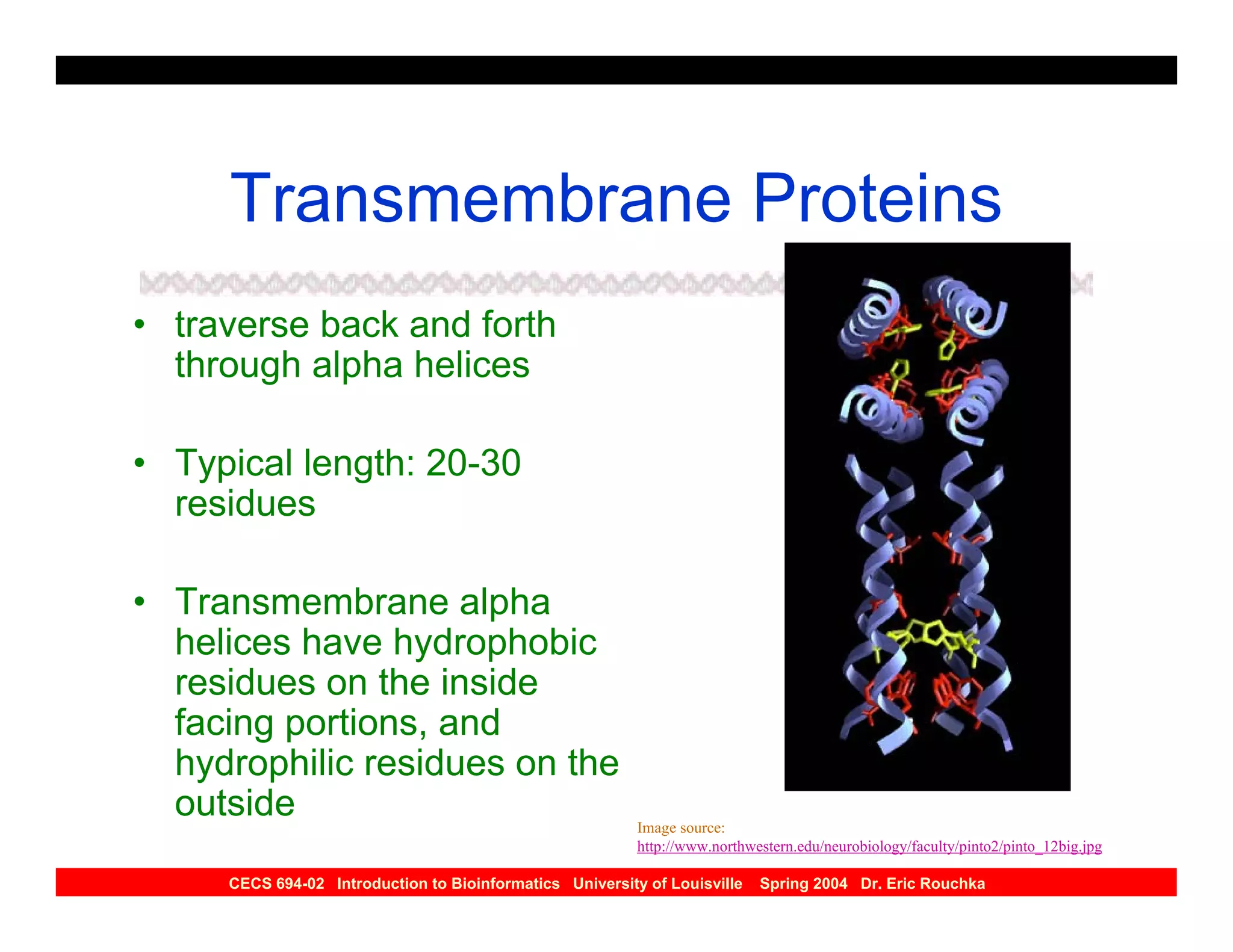
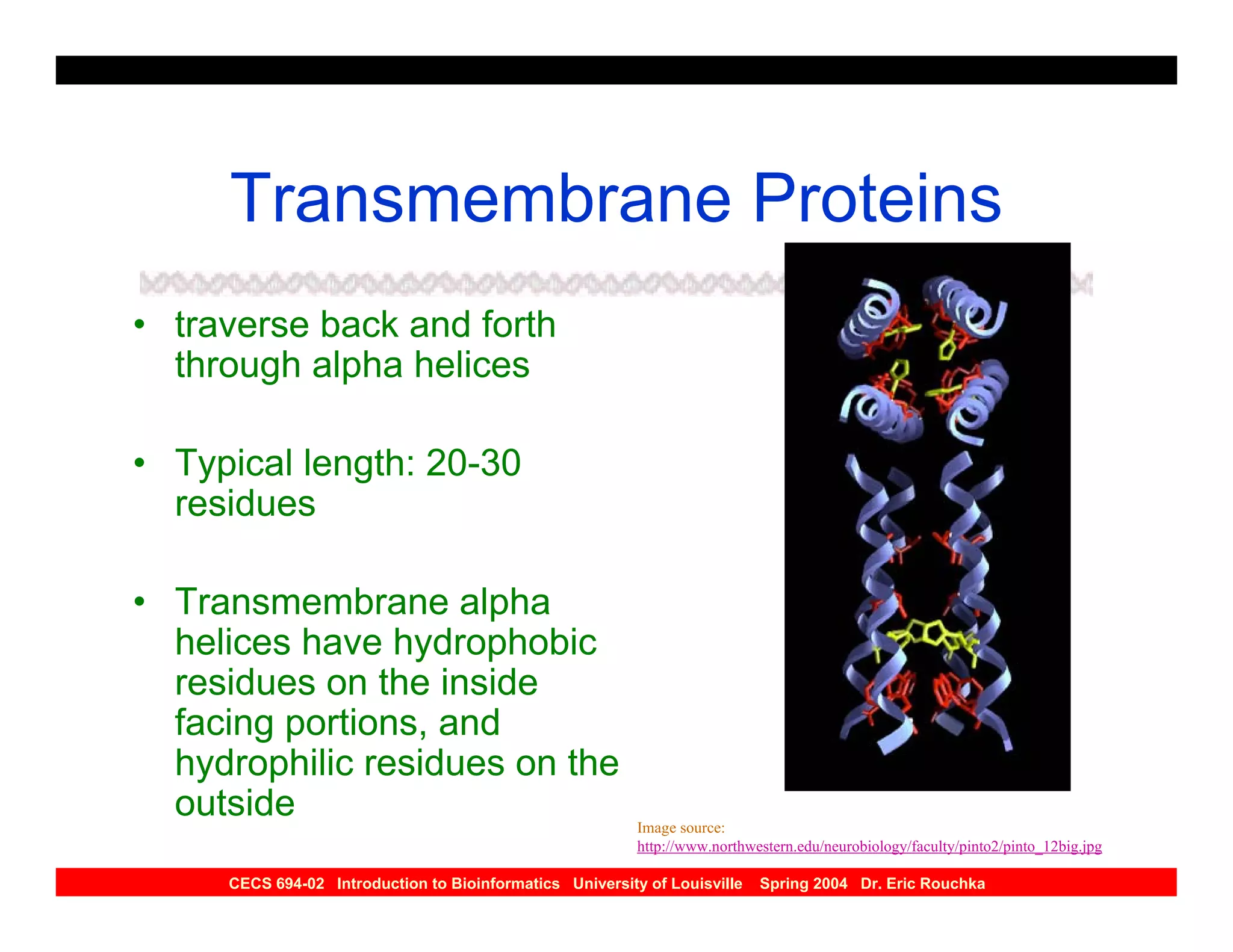
The document discusses protein structure prediction. It begins by reviewing protein structure, including primary, secondary, tertiary, and quaternary structure. It then describes the building blocks of proteins, amino acids, and how their properties allow formation of regular secondary structures like alpha helices and beta sheets. The document outlines different types of secondary structure and how their patterns of hydrogen bonding influence 3D structure. It concludes by describing six classes of protein structure defined by their arrangements of alpha helices and beta sheets.