Downloaded 262 times

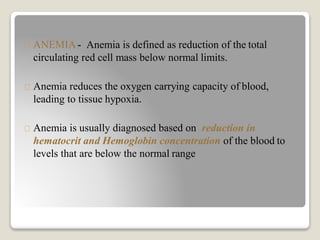
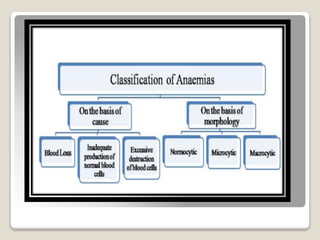



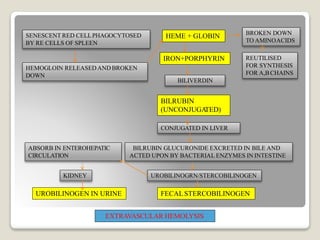
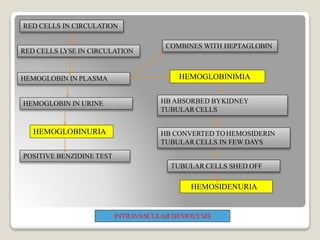
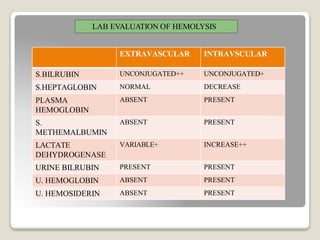
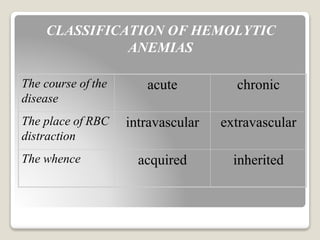
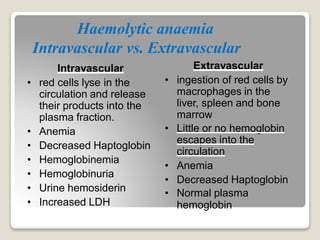







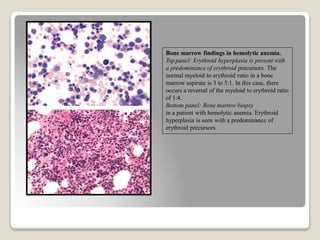










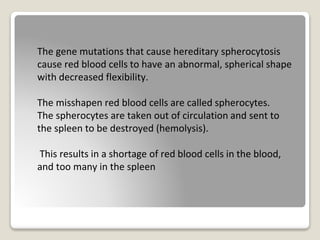


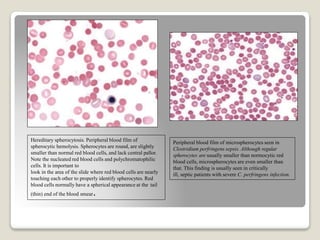



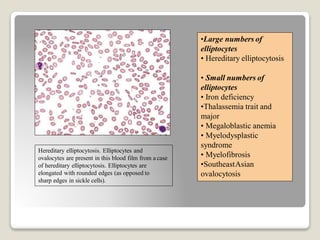










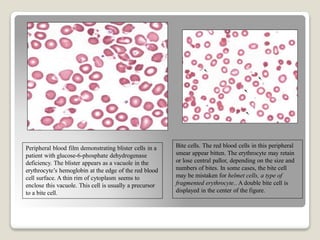
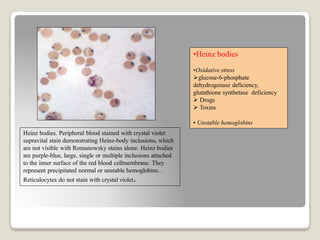

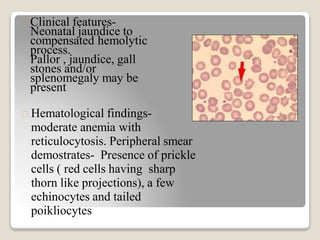





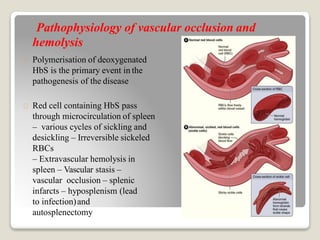








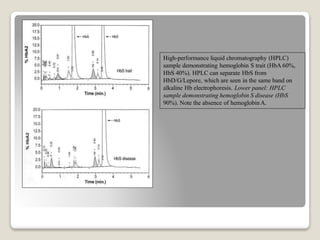







Hereditary spherocytosis is a hereditary hemolytic anemia caused by a red blood cell membrane defect that results in spherocytosis. The misshapen red blood cells, called spherocytes, are destroyed by the spleen, leading to hemolysis and a shortage of red blood cells. Clinical features include jaundice, splenomegaly, and gallstones. Laboratory findings show microspherocytes on peripheral blood film and increased reticulocytes, with normal red blood cell size.


































































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)











