Downloaded 476 times
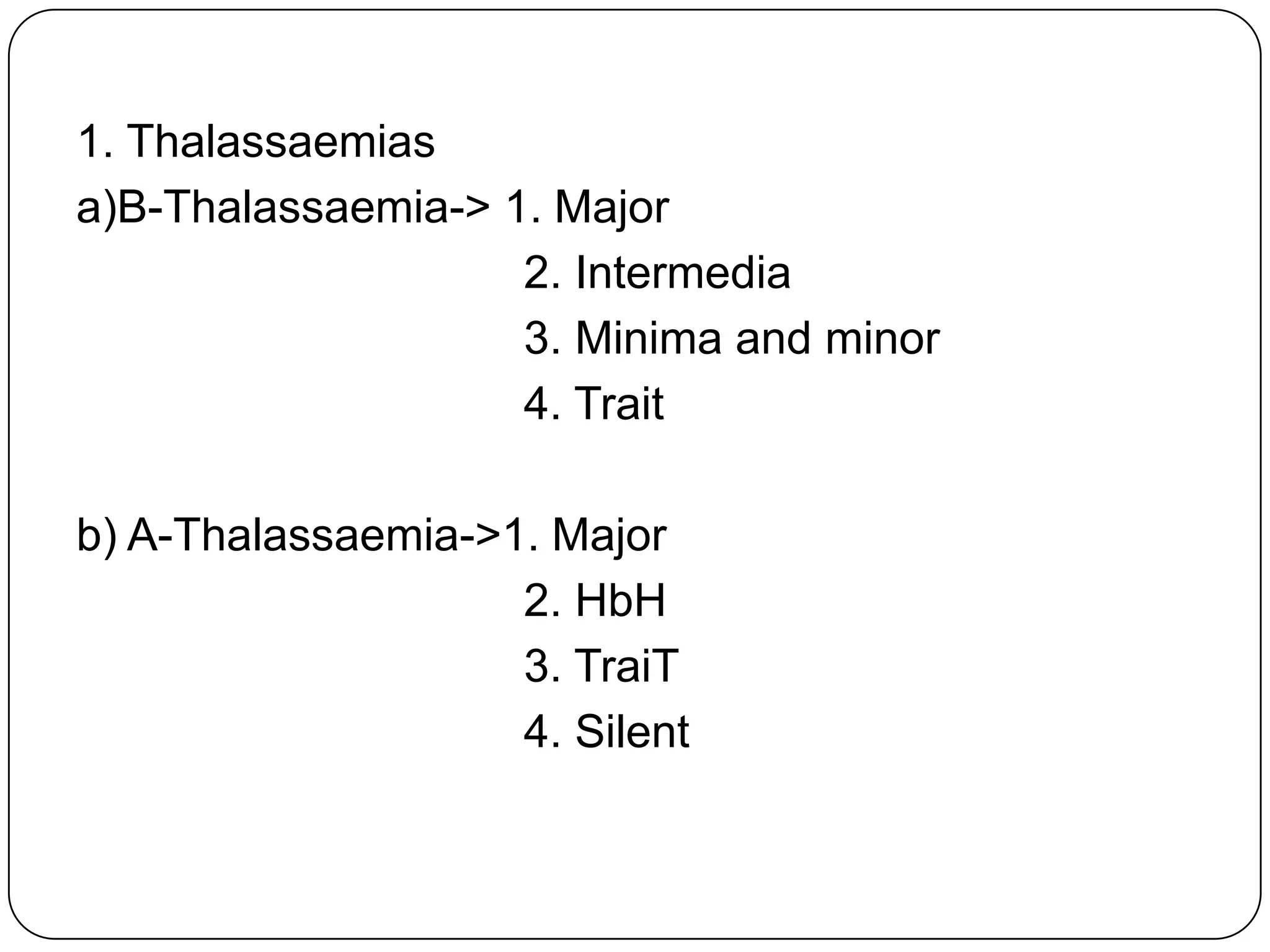



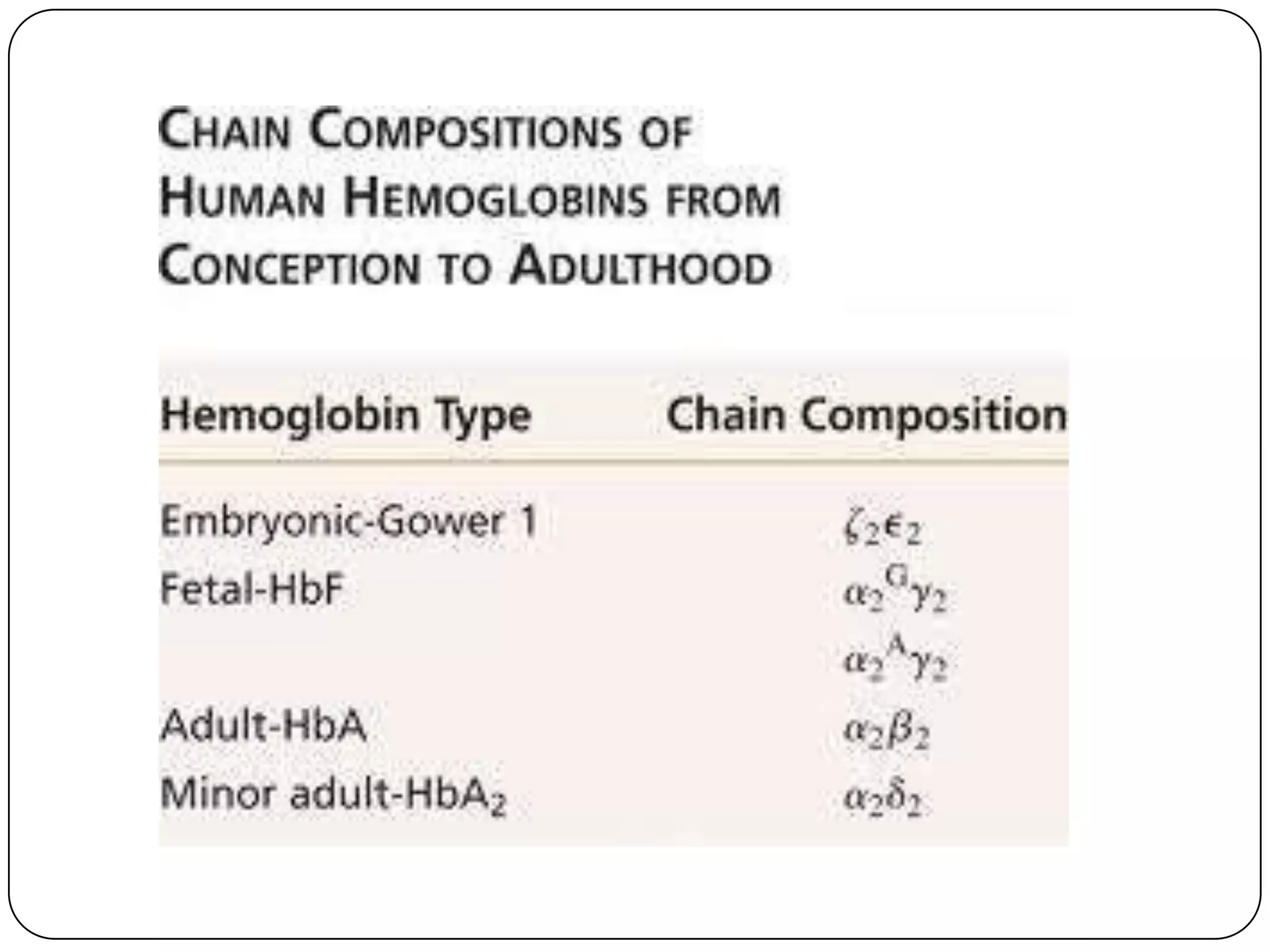
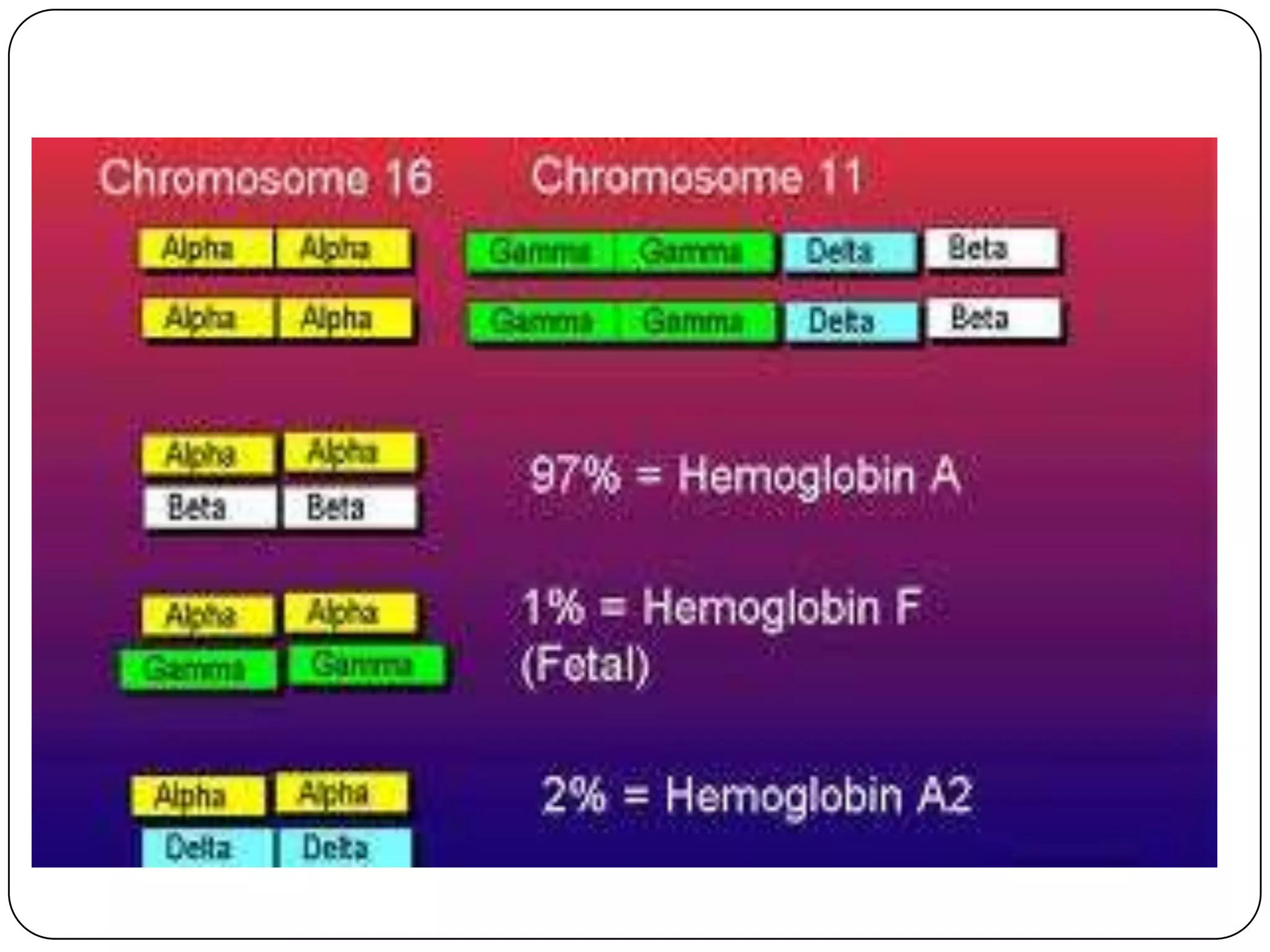

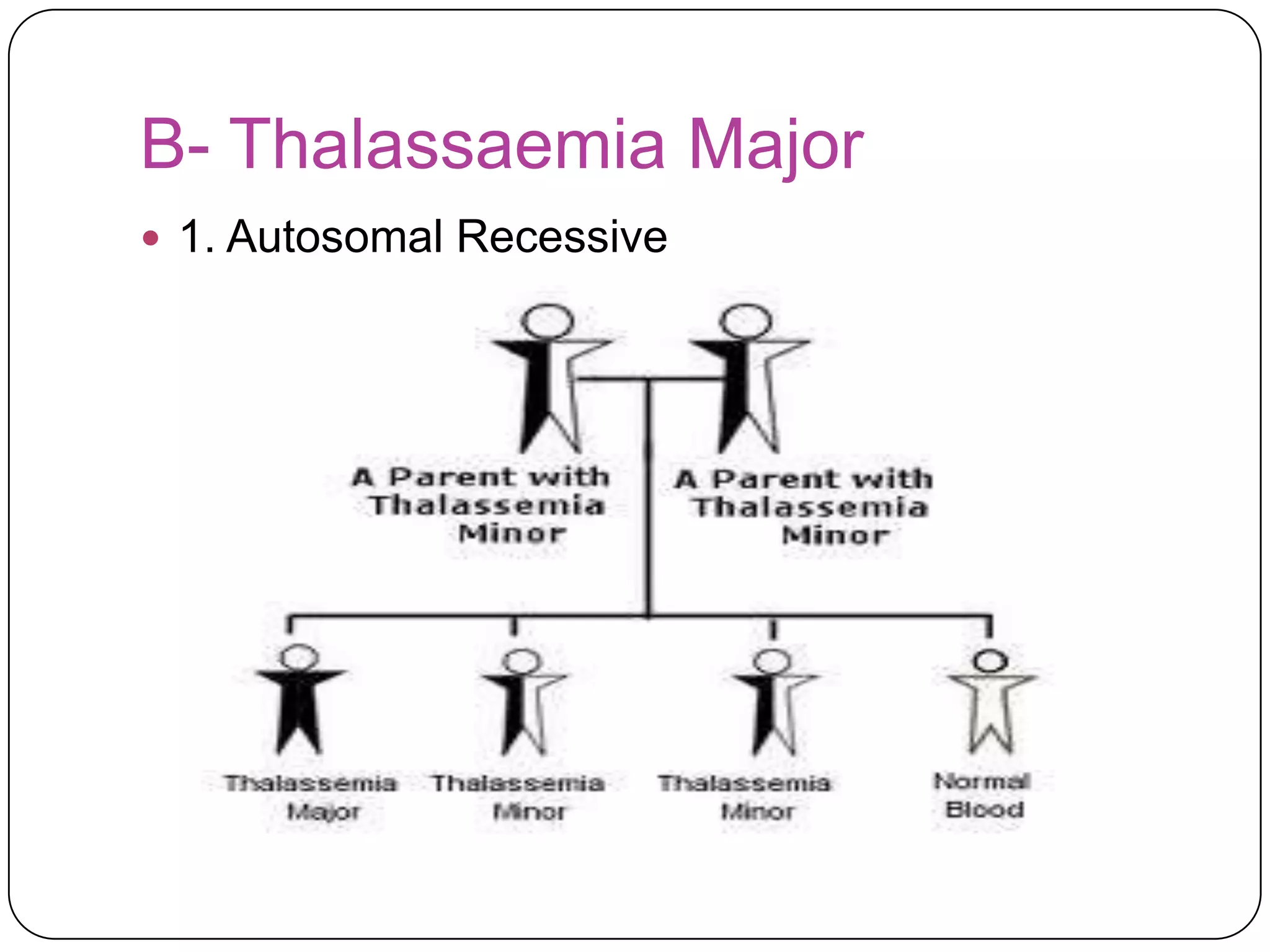
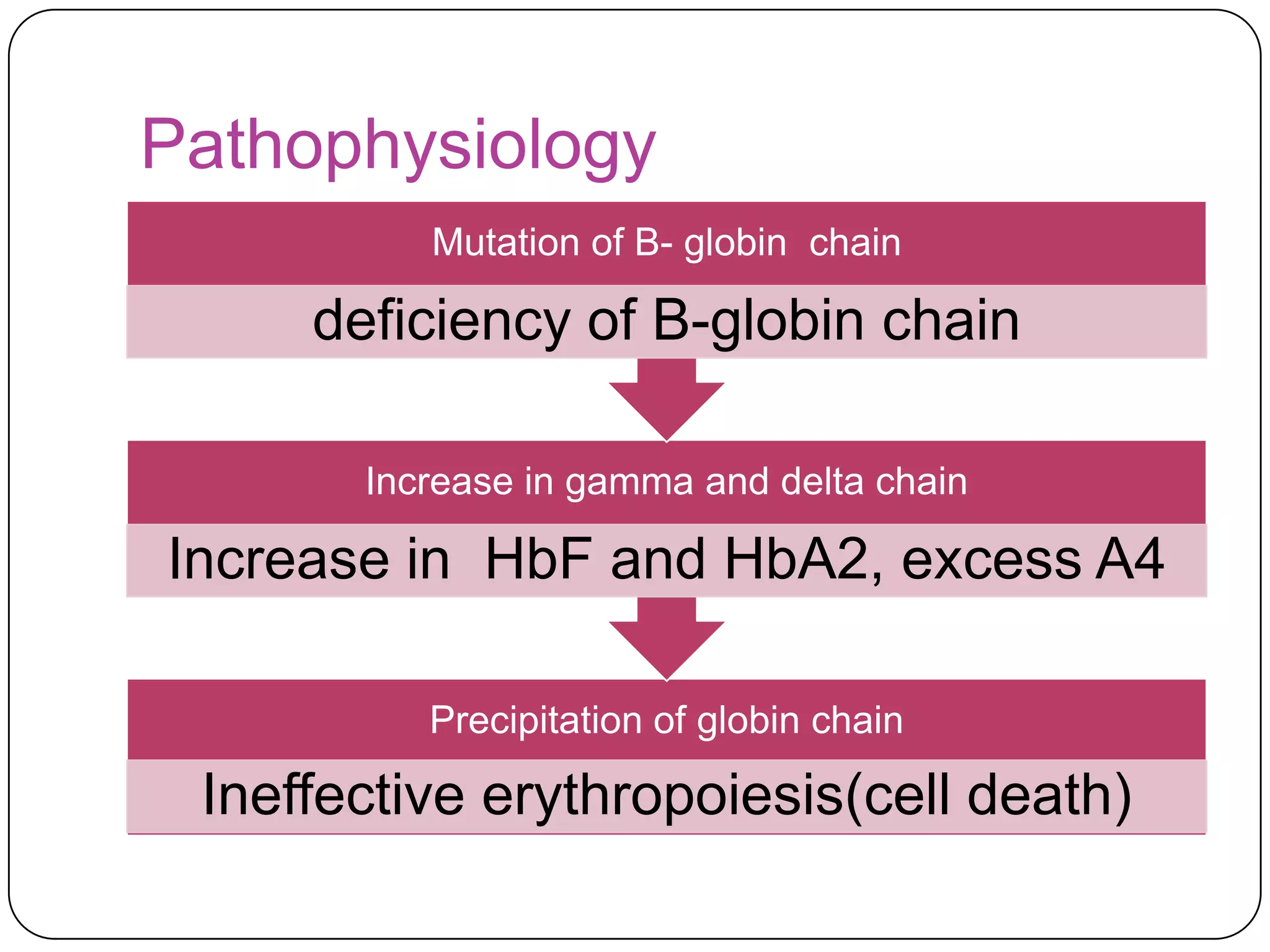


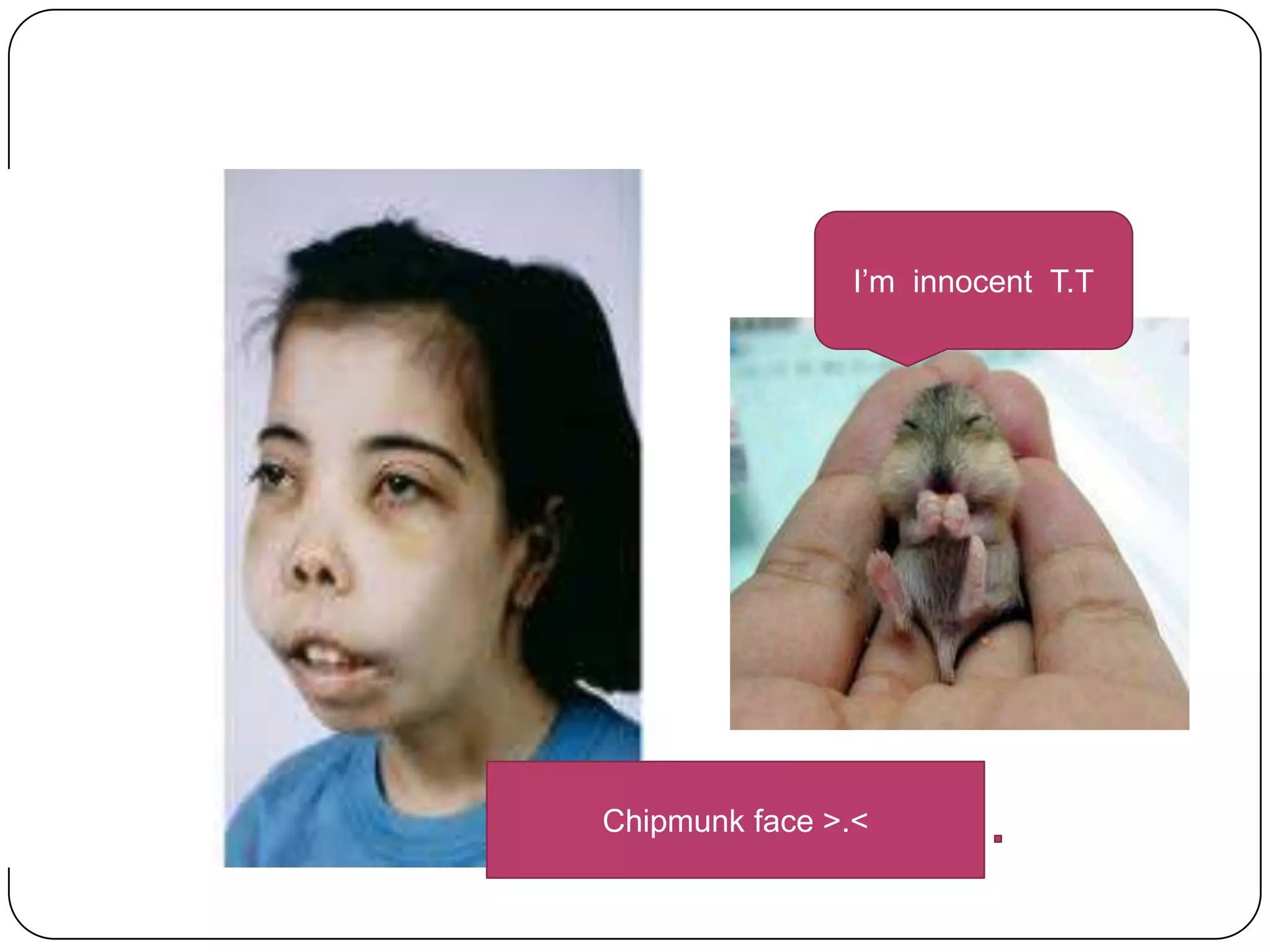
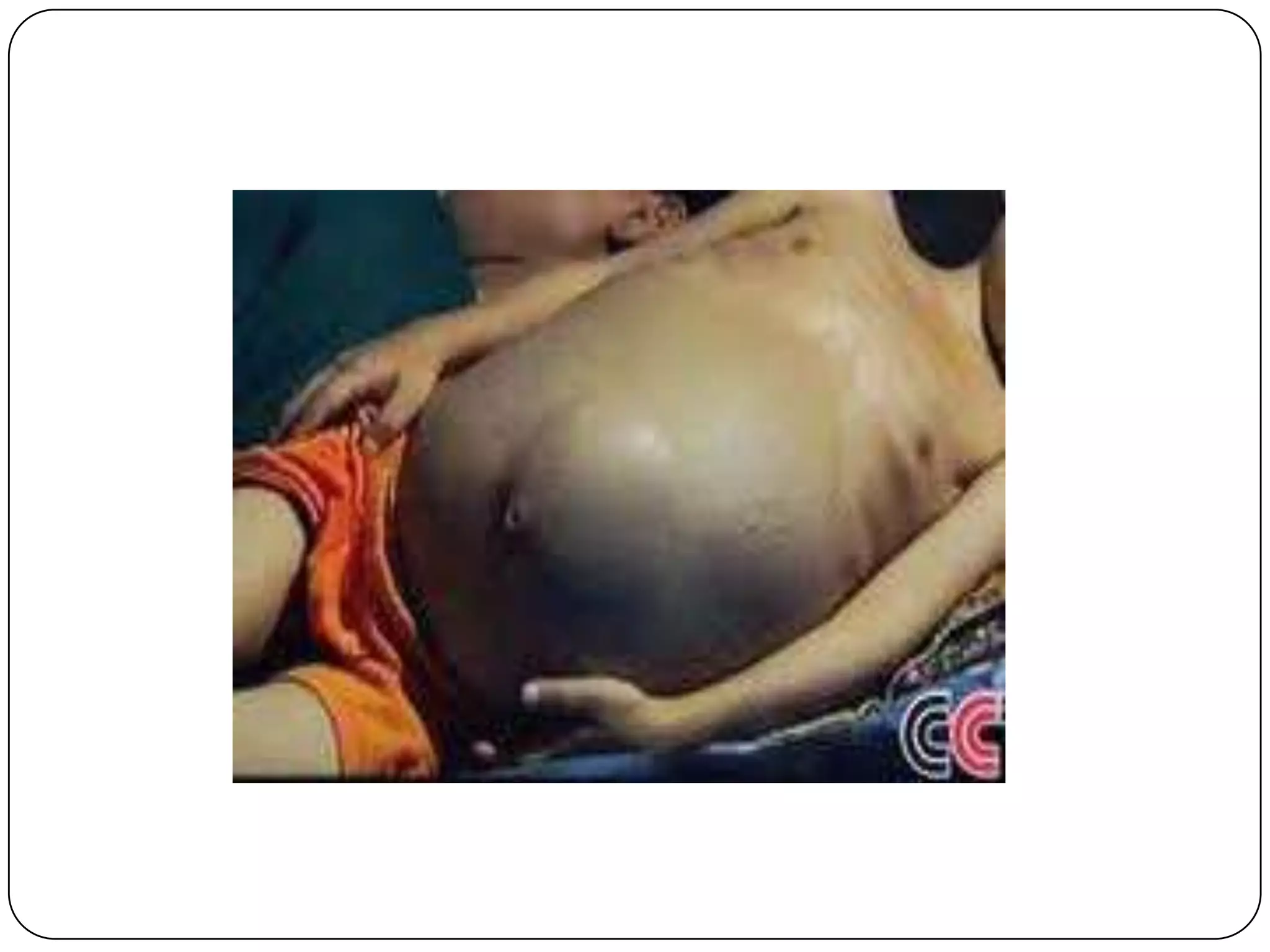


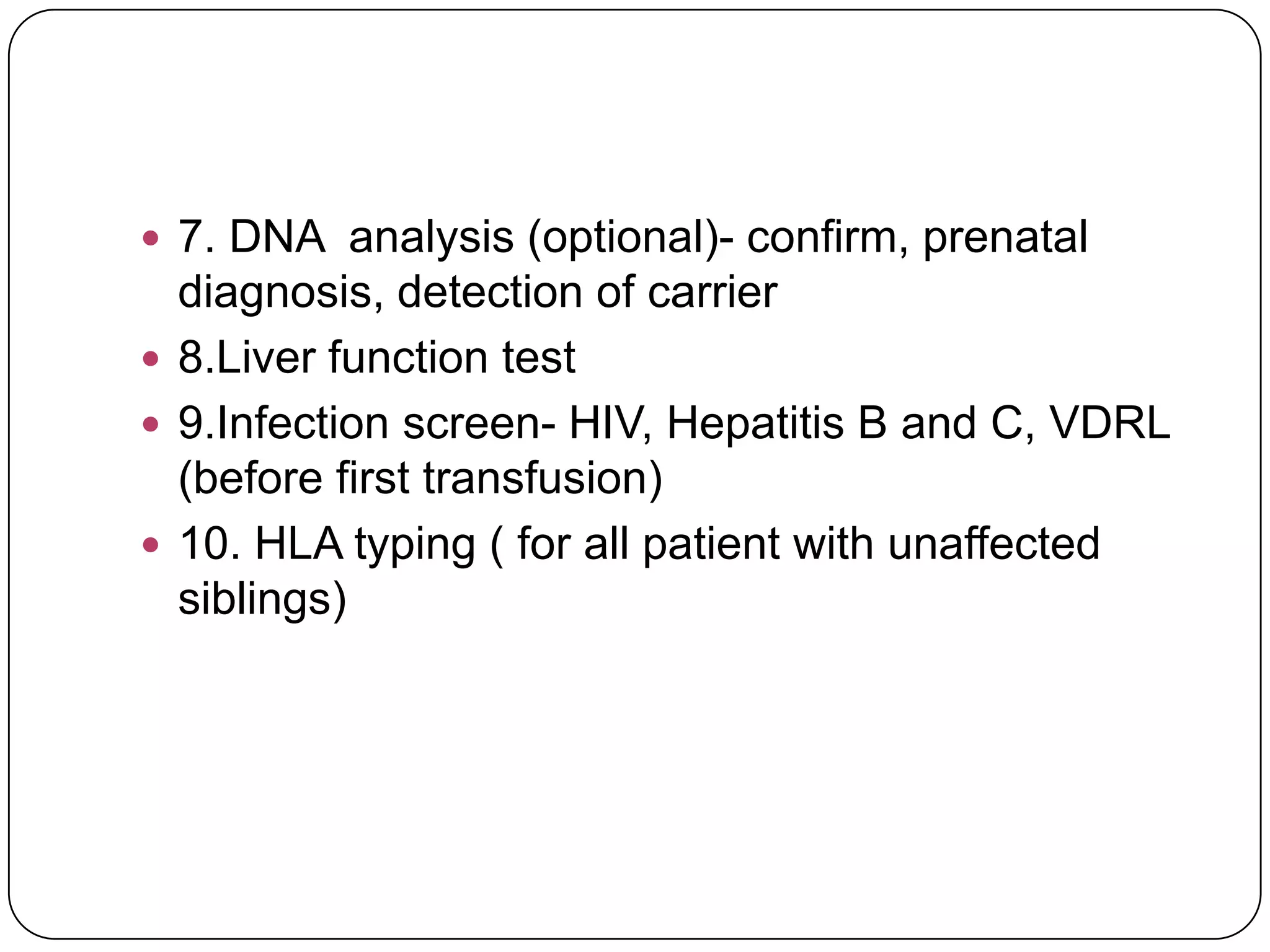
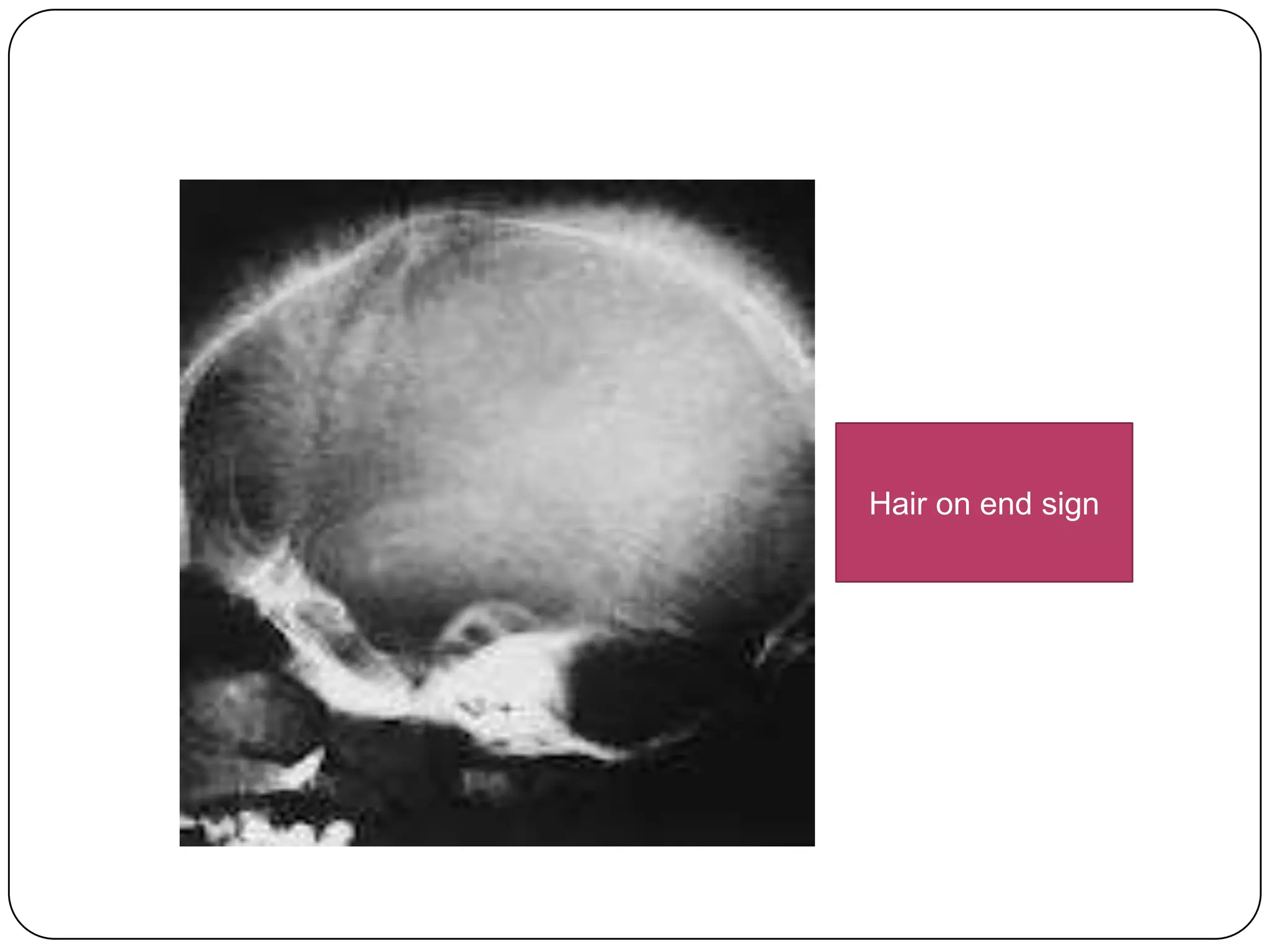
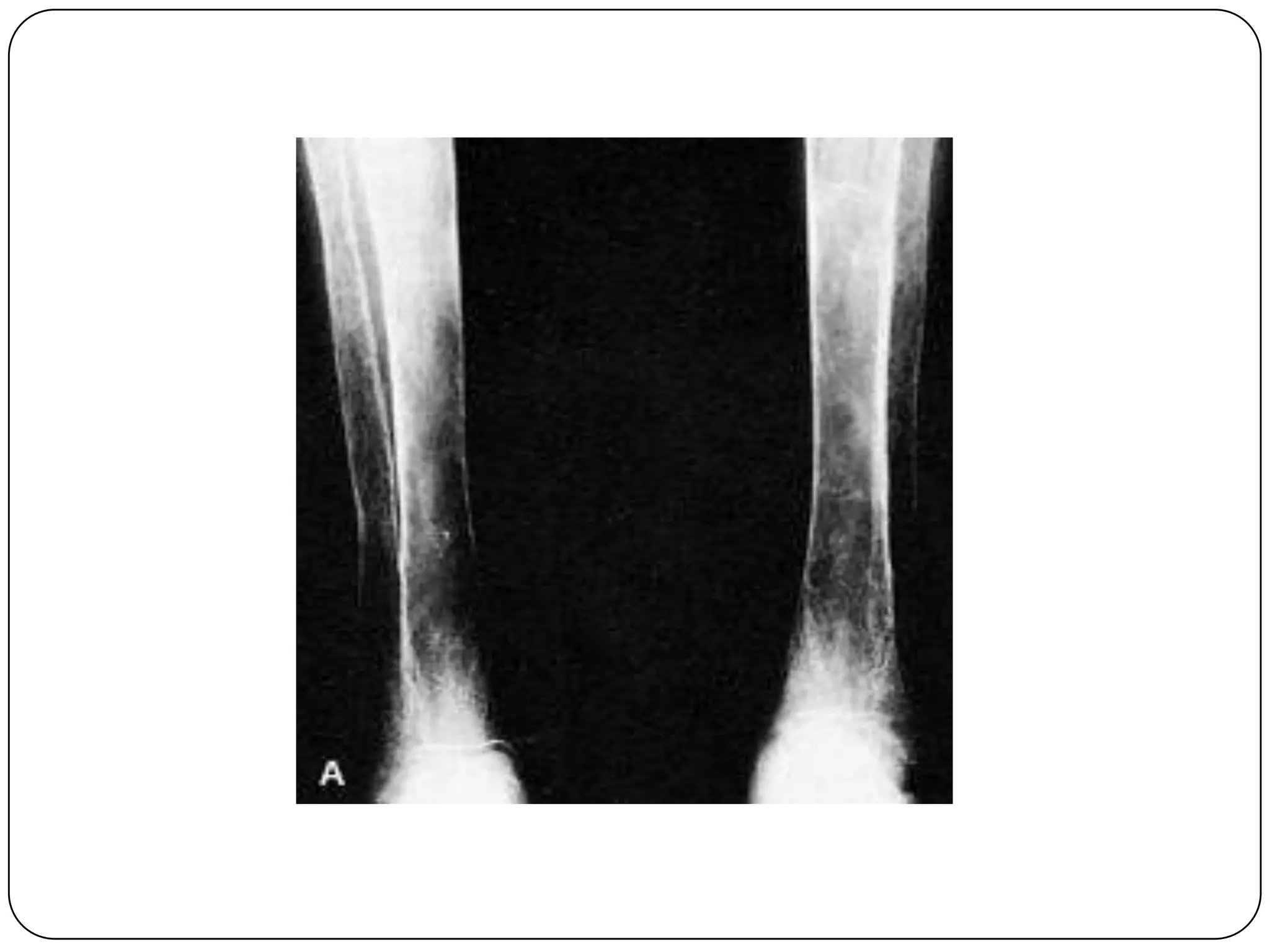
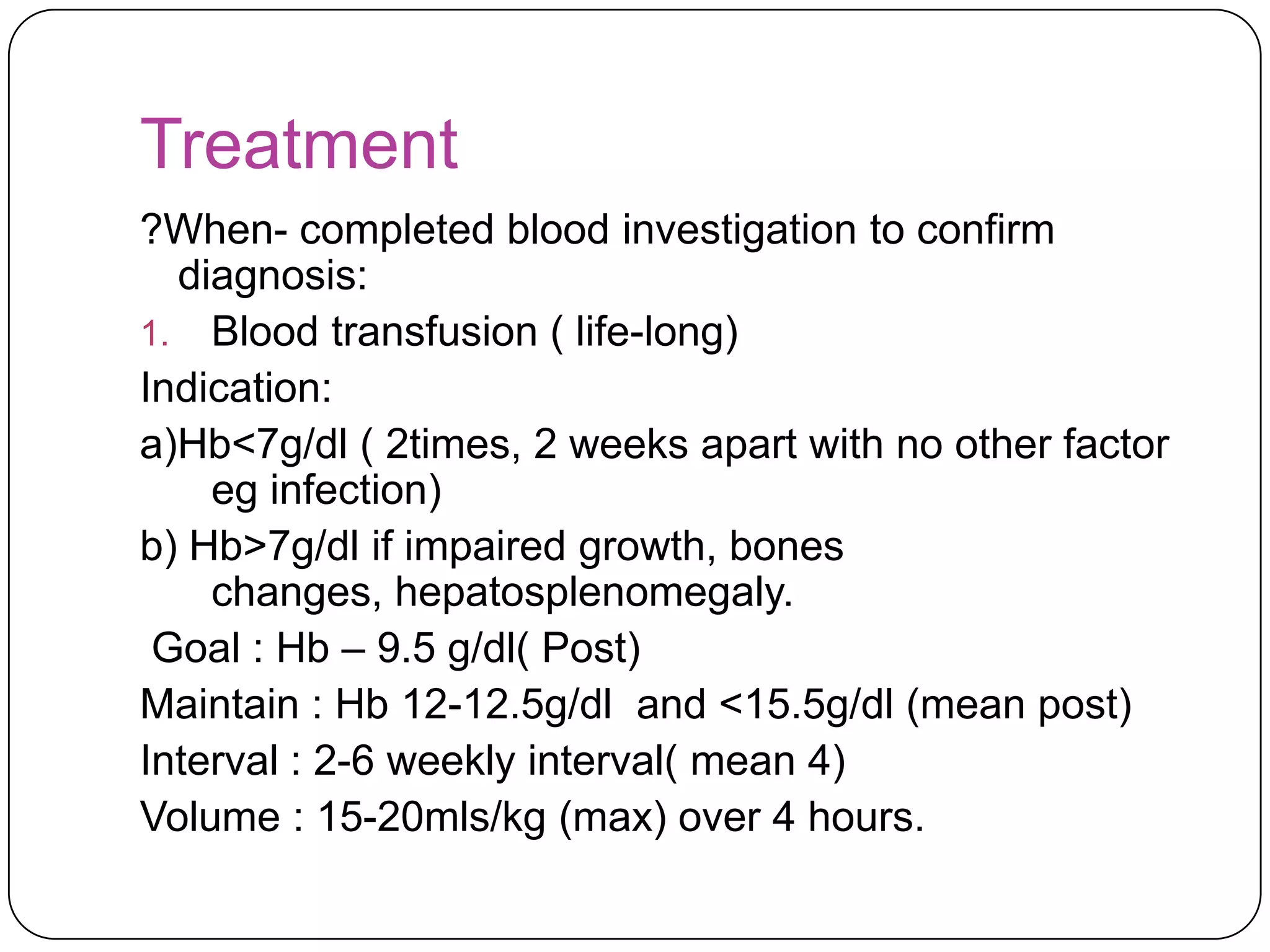
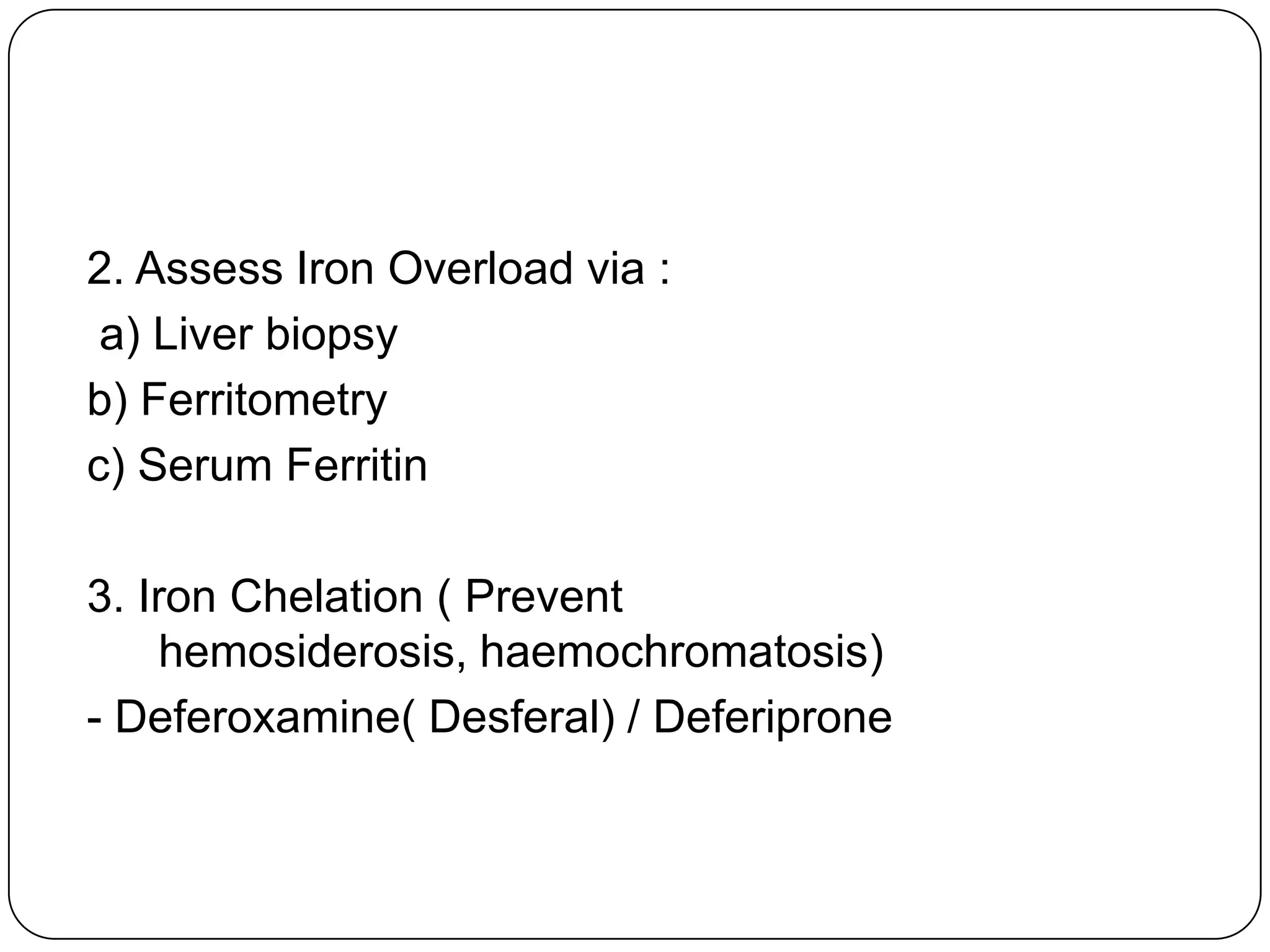

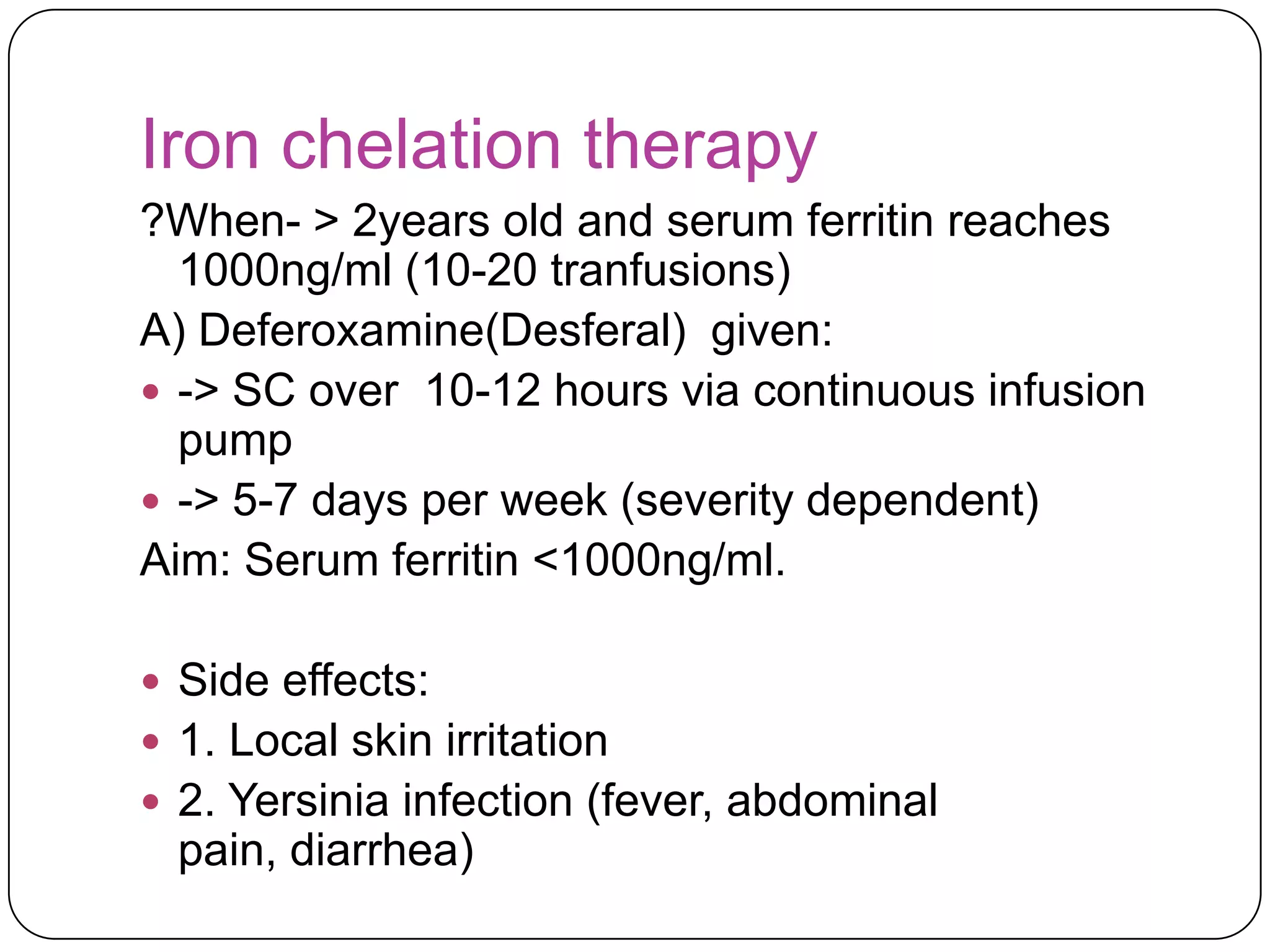
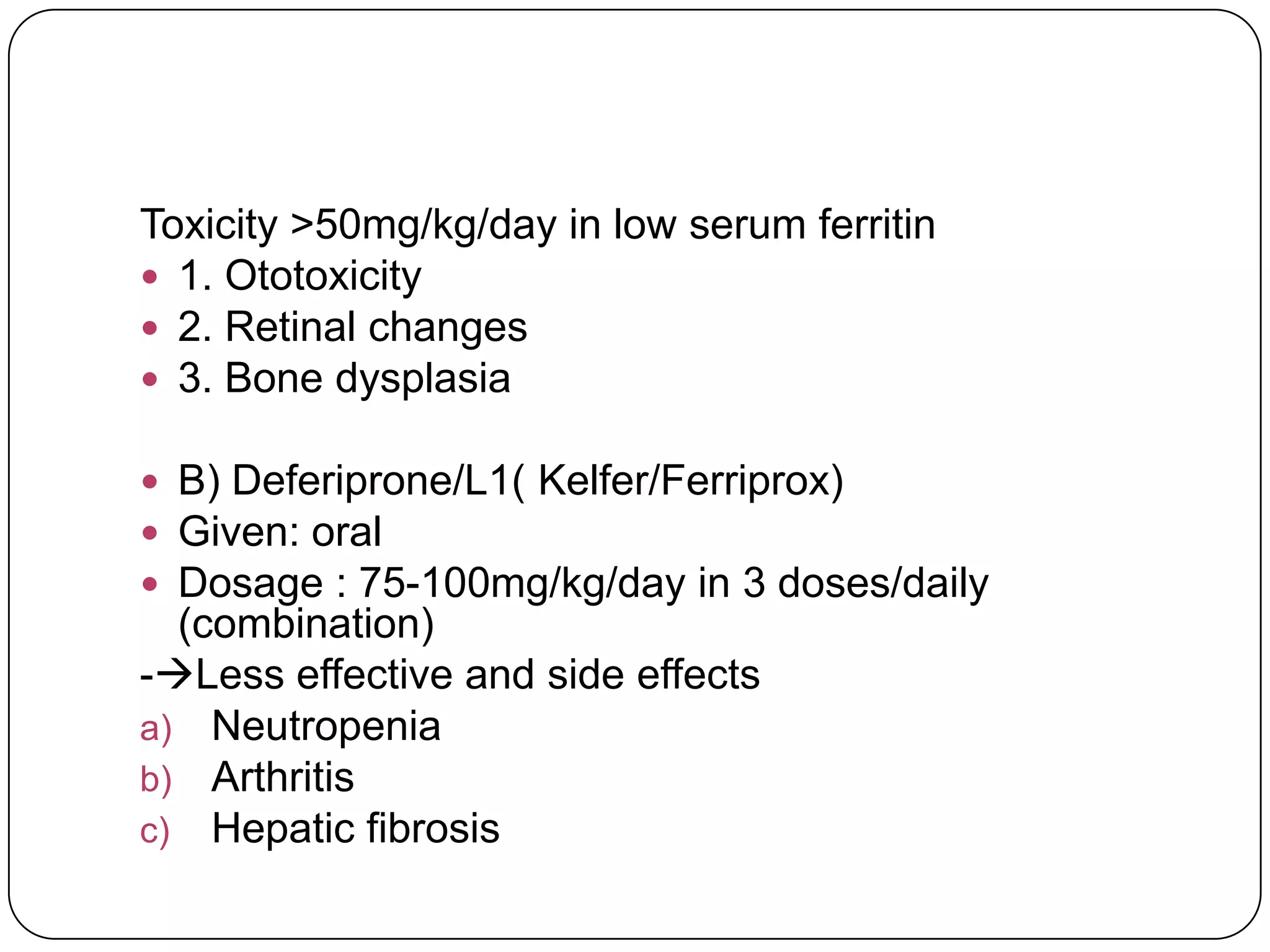







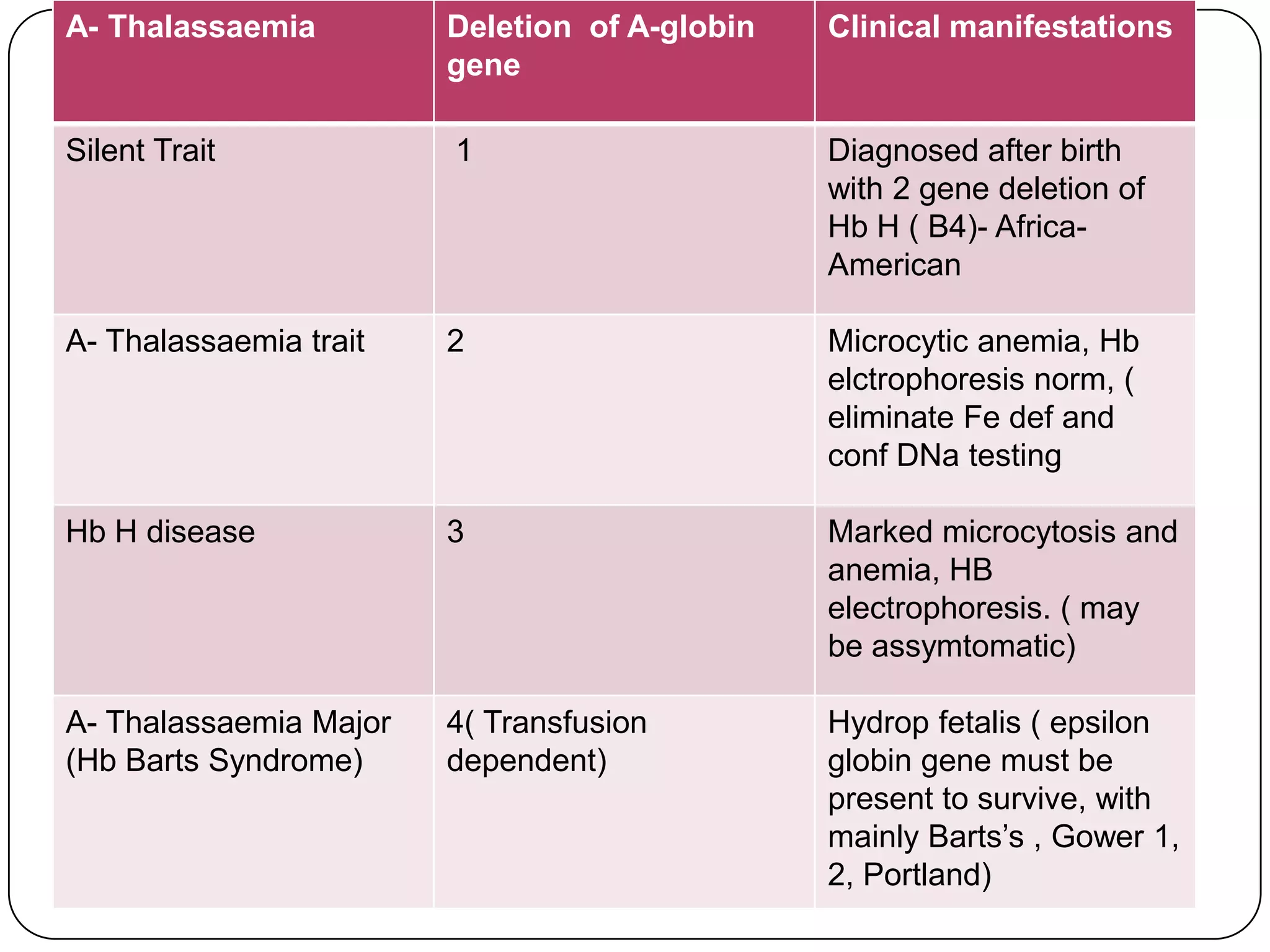
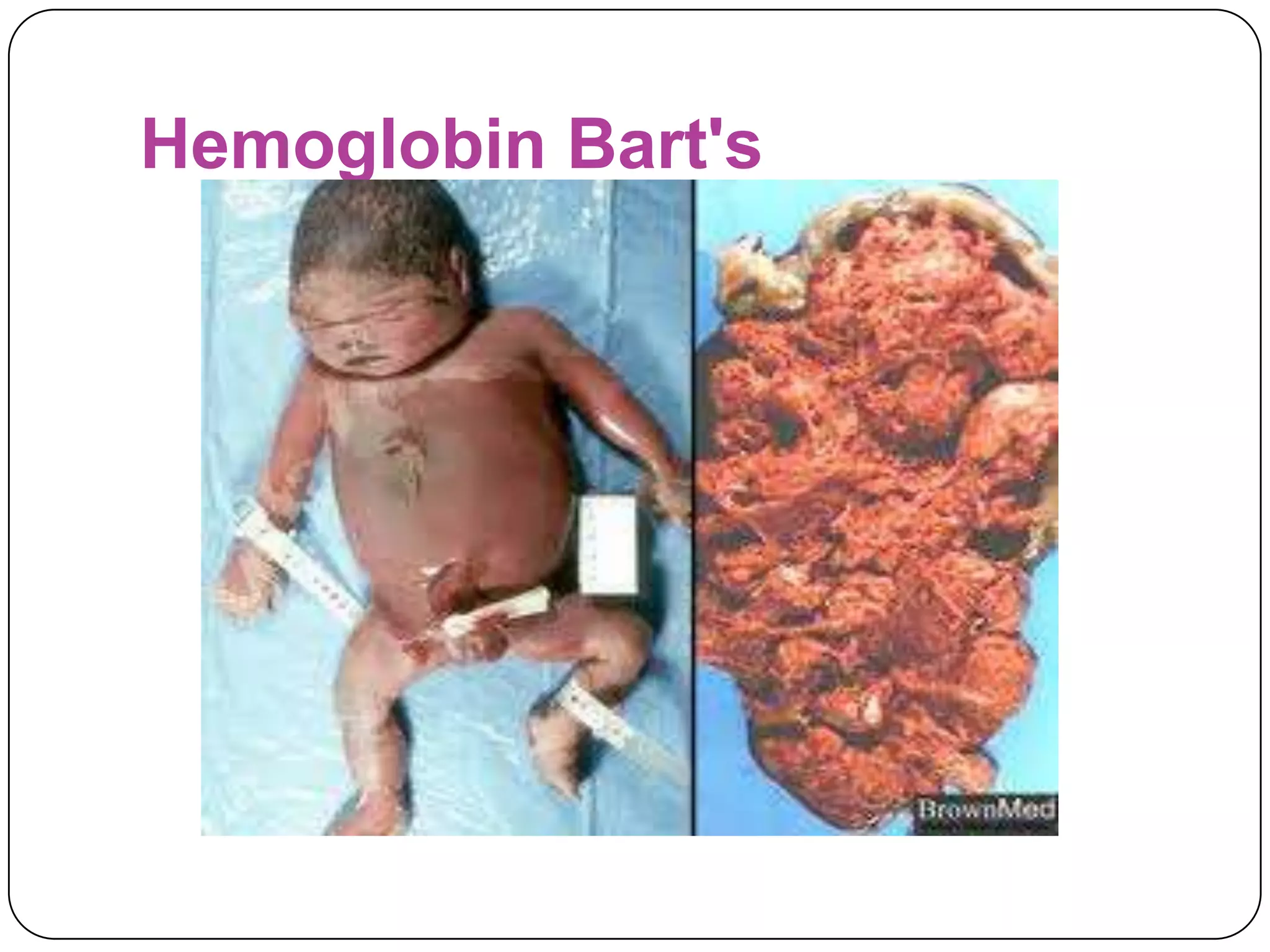

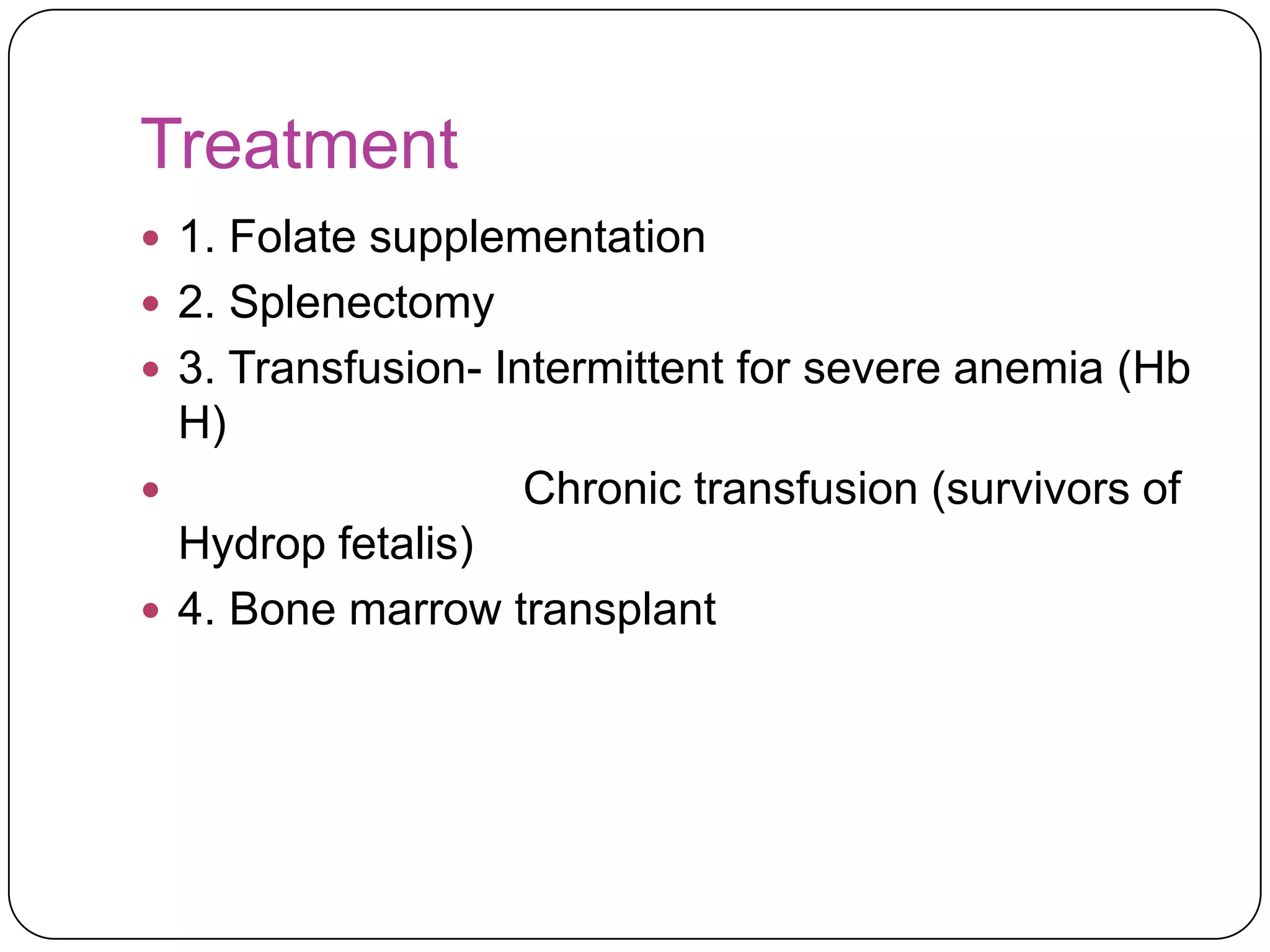


Hemolytic anemia can be caused by thalassemias, the most common genetic blood disorders worldwide characterized by reduced or absent globin chain production leading to anemia. The main types are beta thalassemia major requiring lifelong blood transfusions, and alpha thalassemia including trait, HbH disease, and rare hydrops fetalis. Treatment focuses on blood transfusions, iron chelation therapy, and in severe cases bone marrow transplantation.






























































































