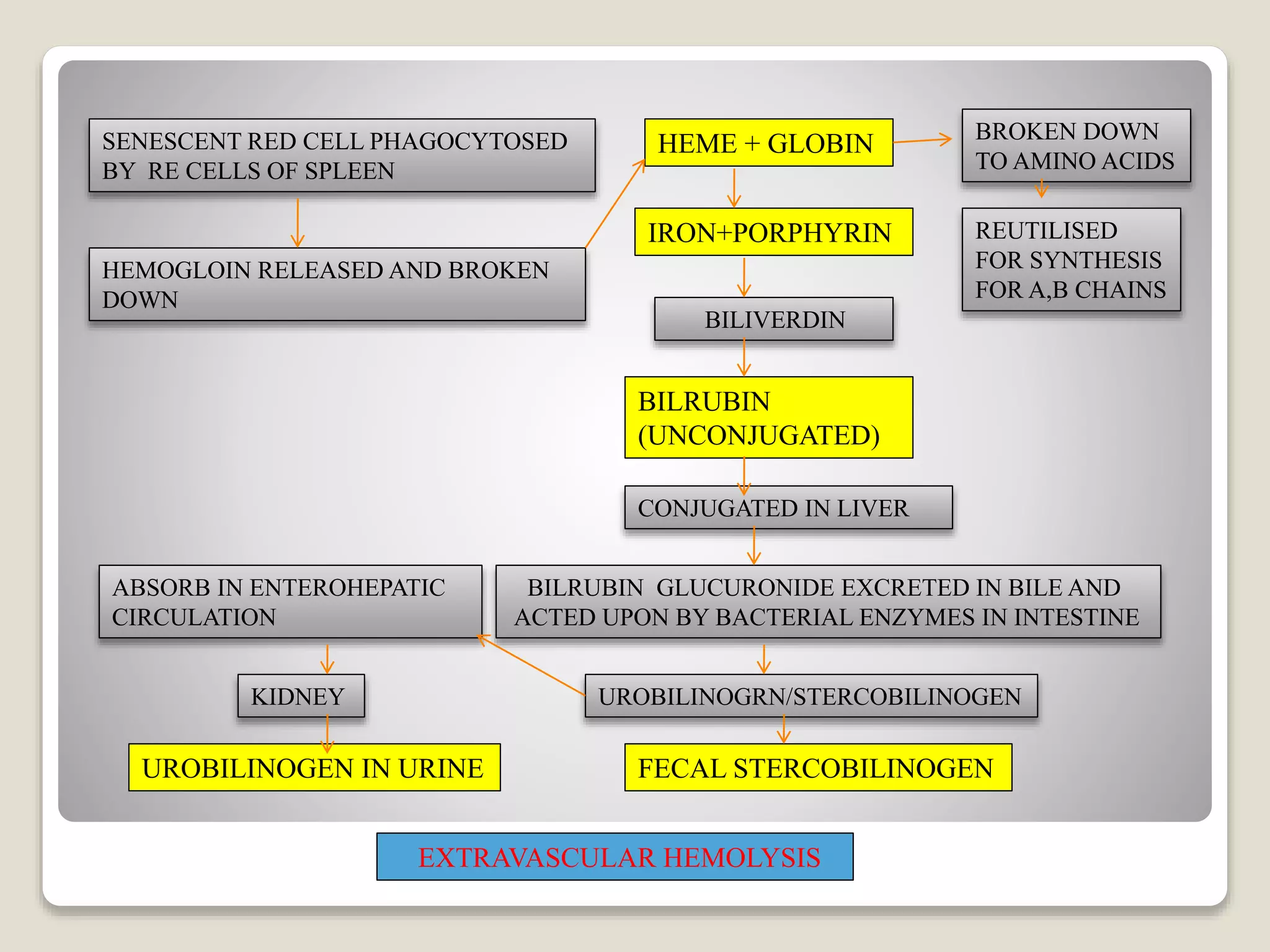
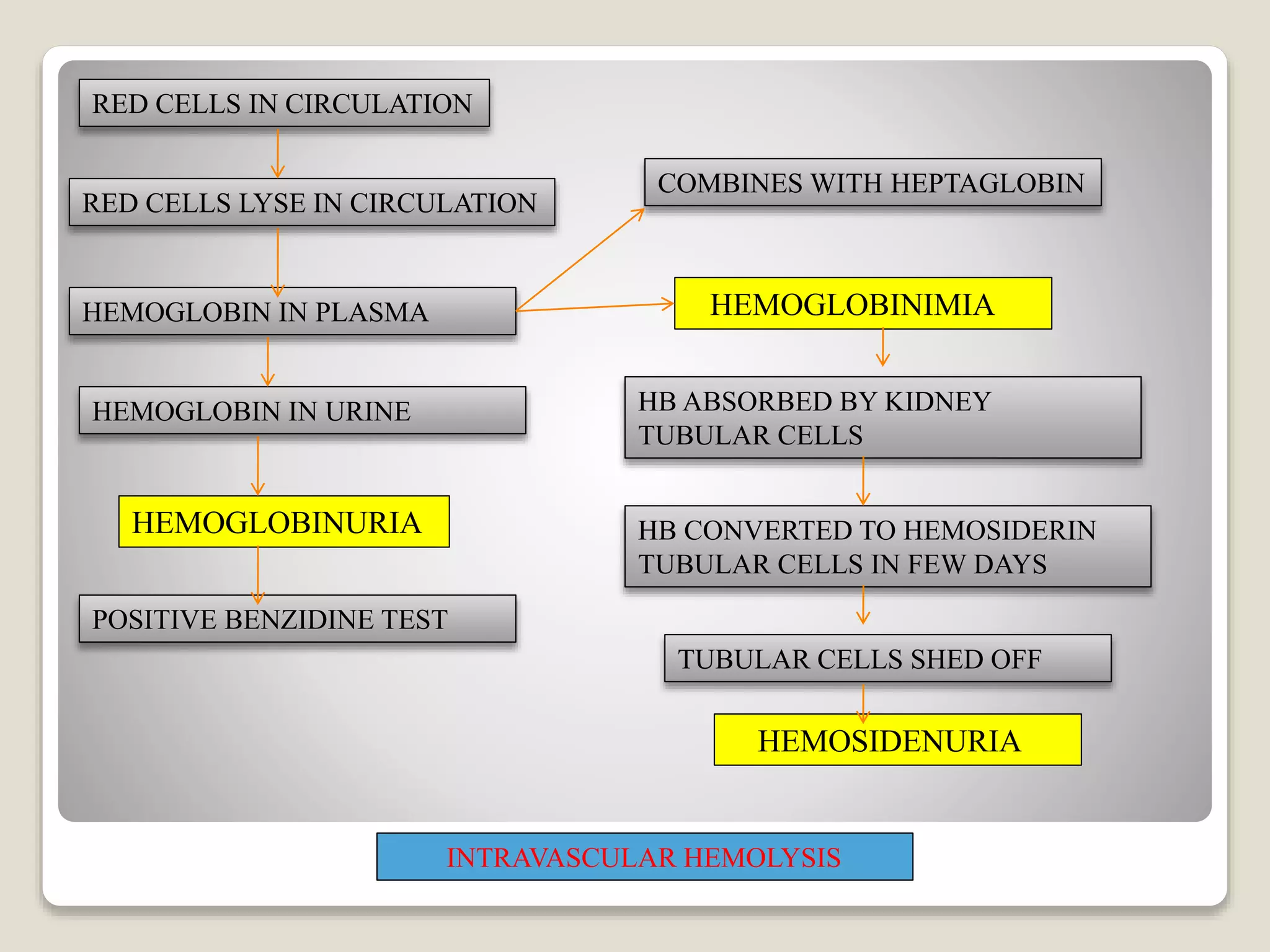
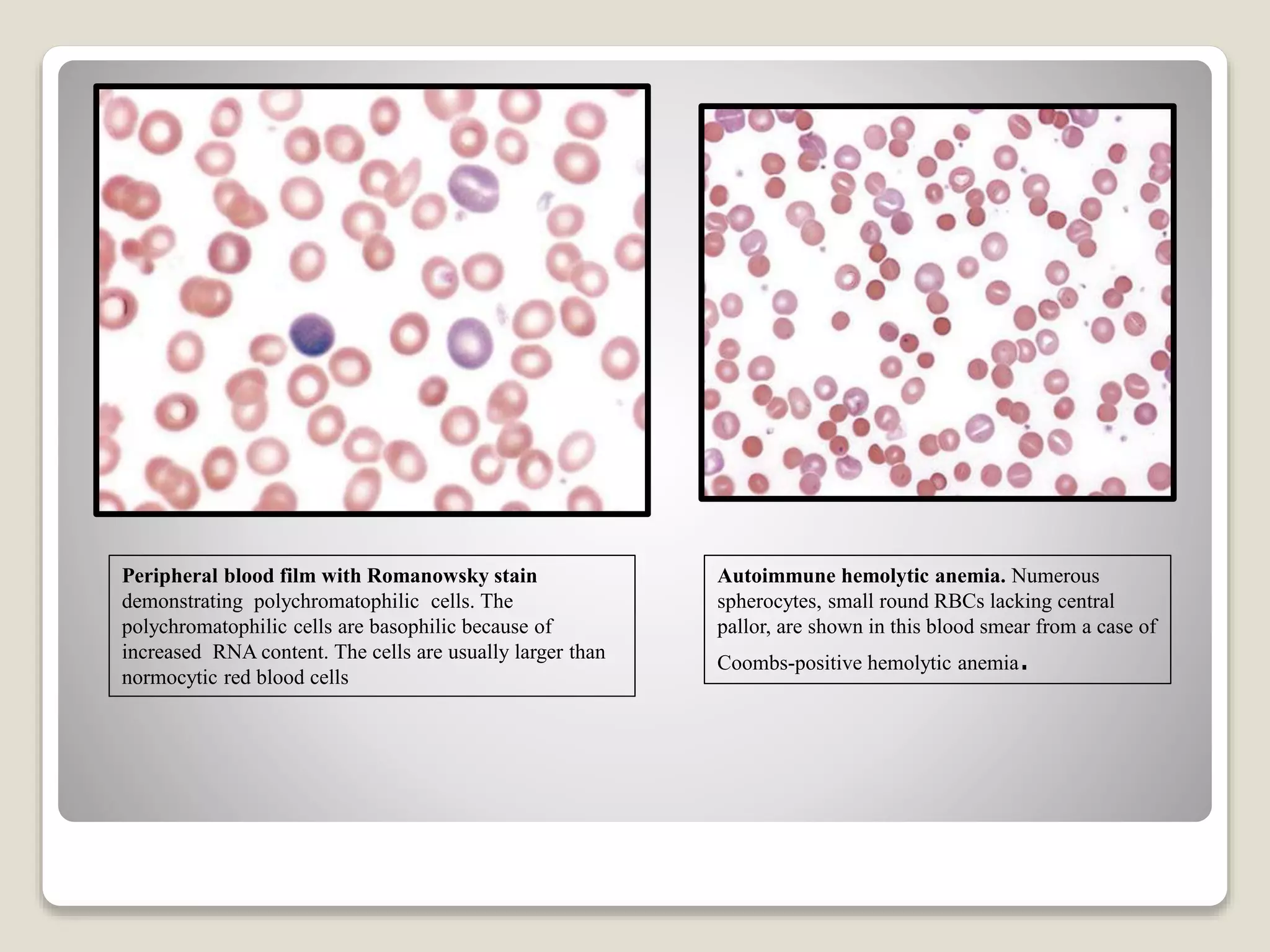
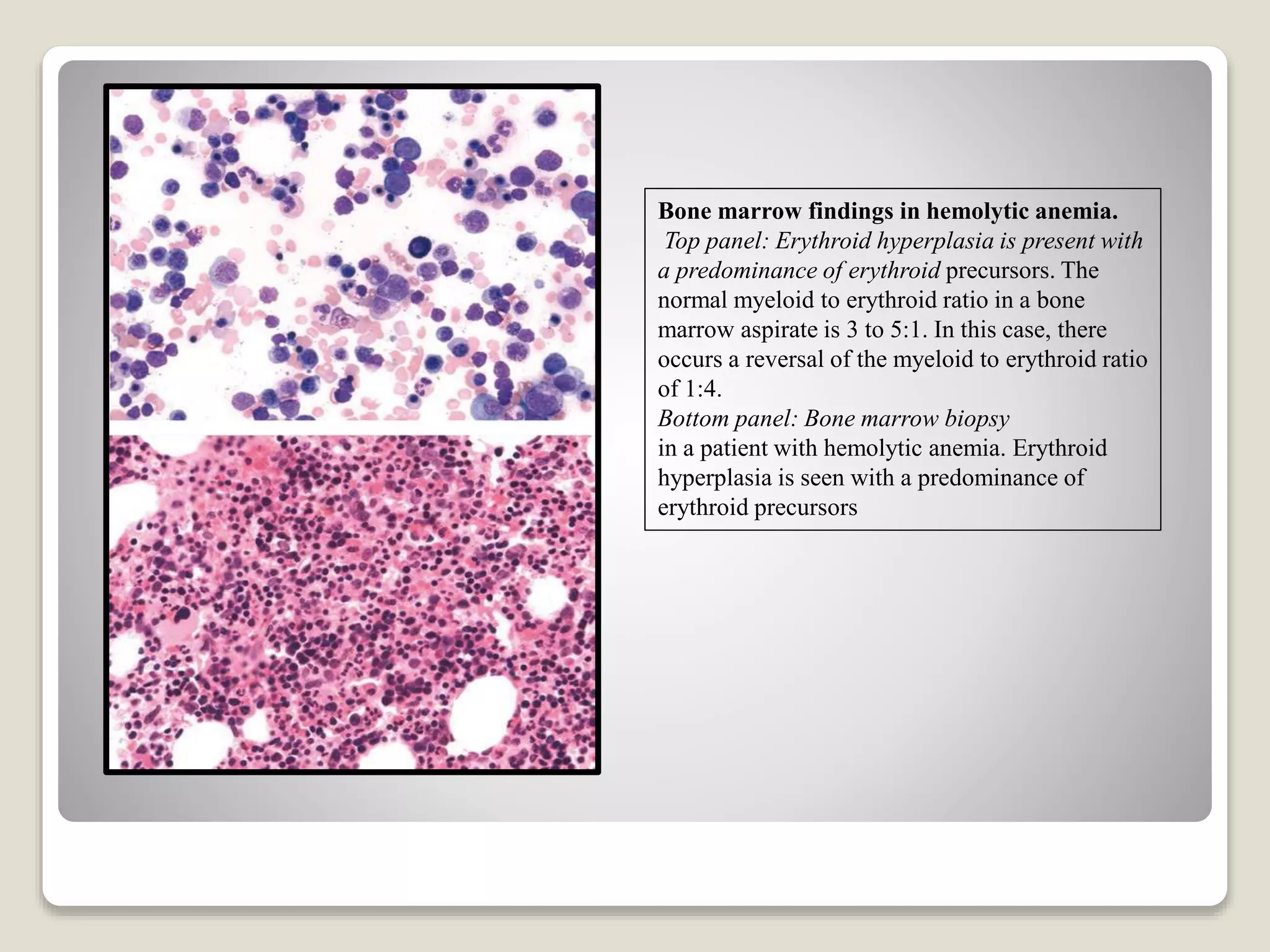
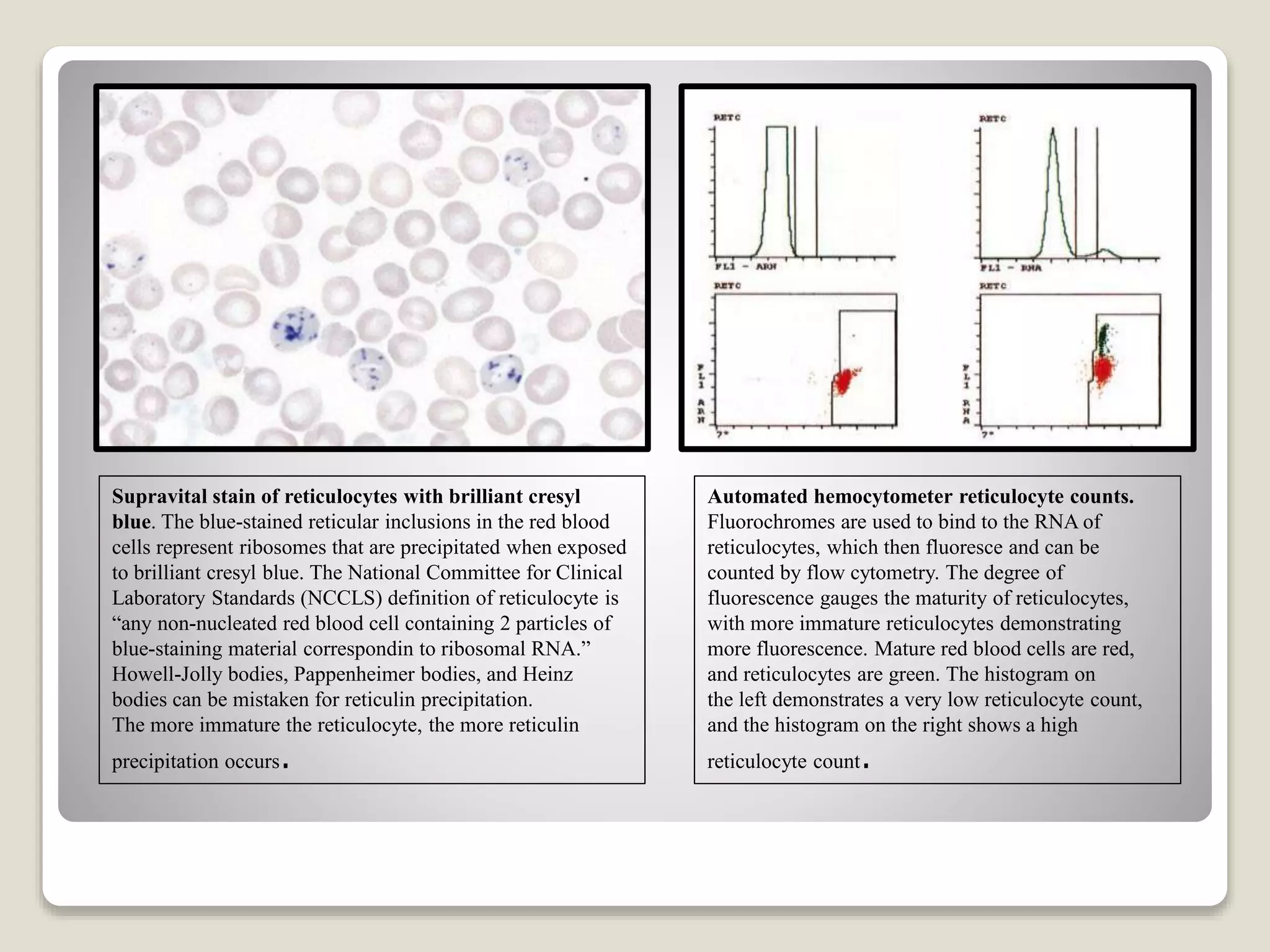
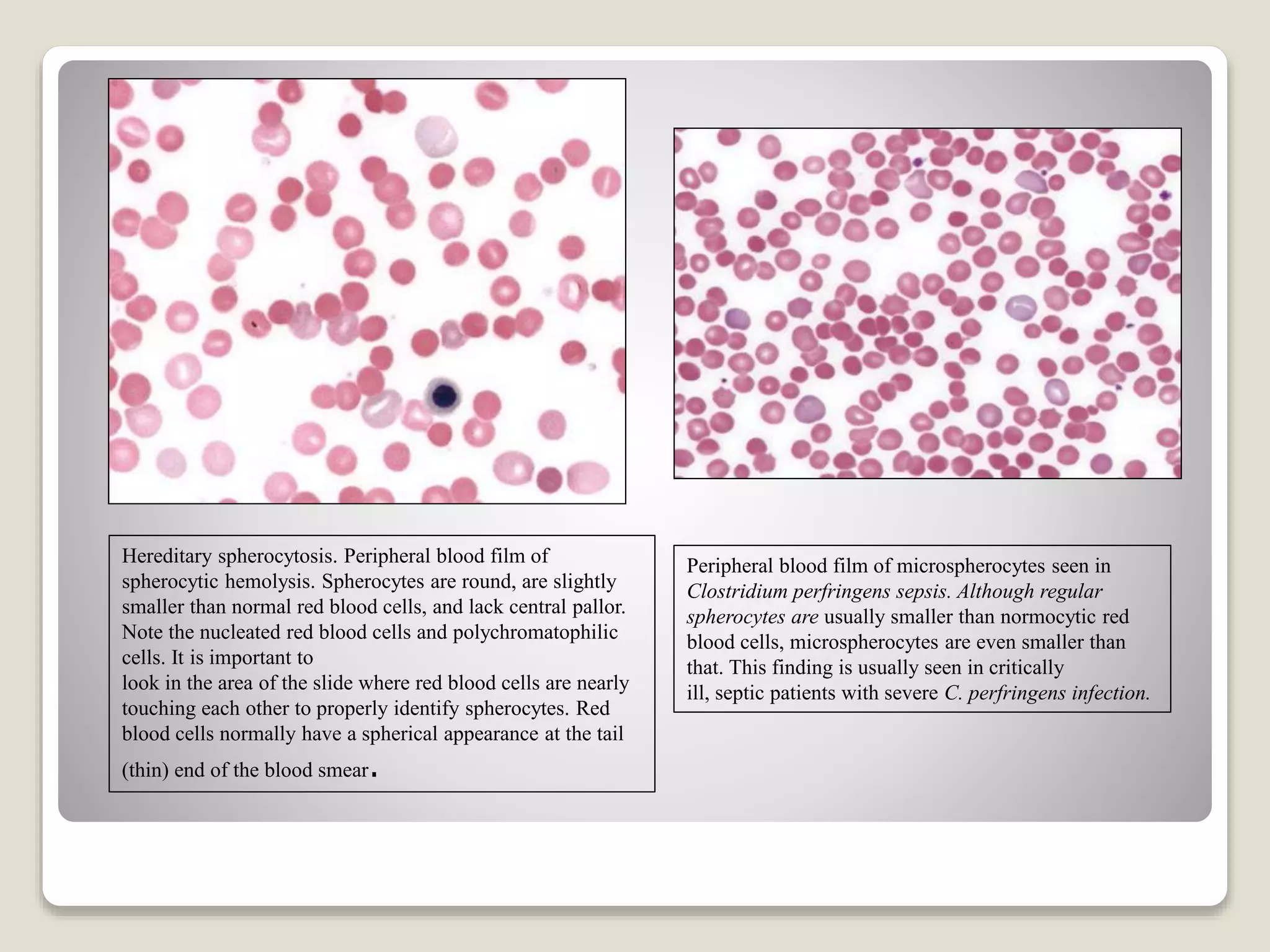
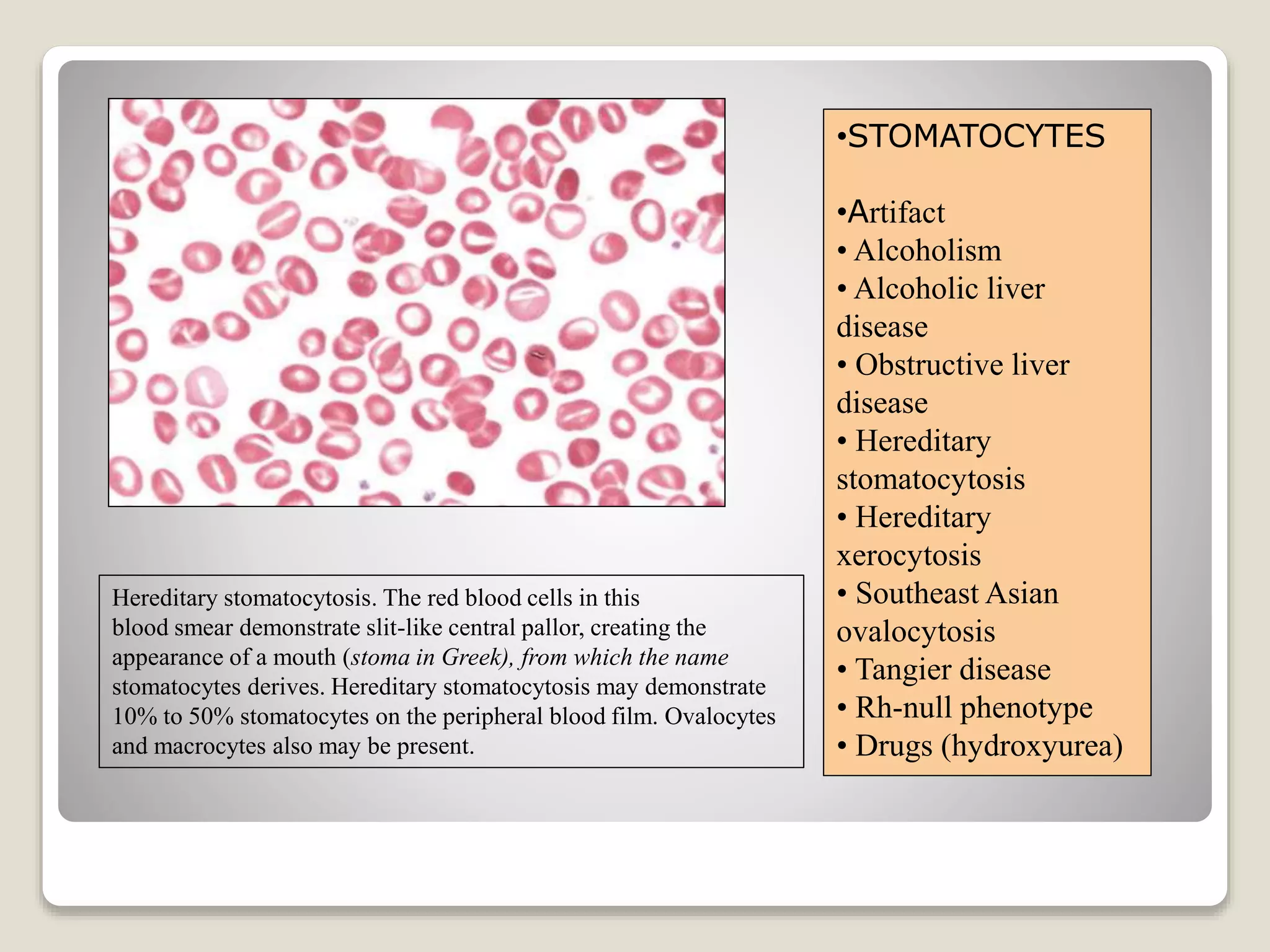
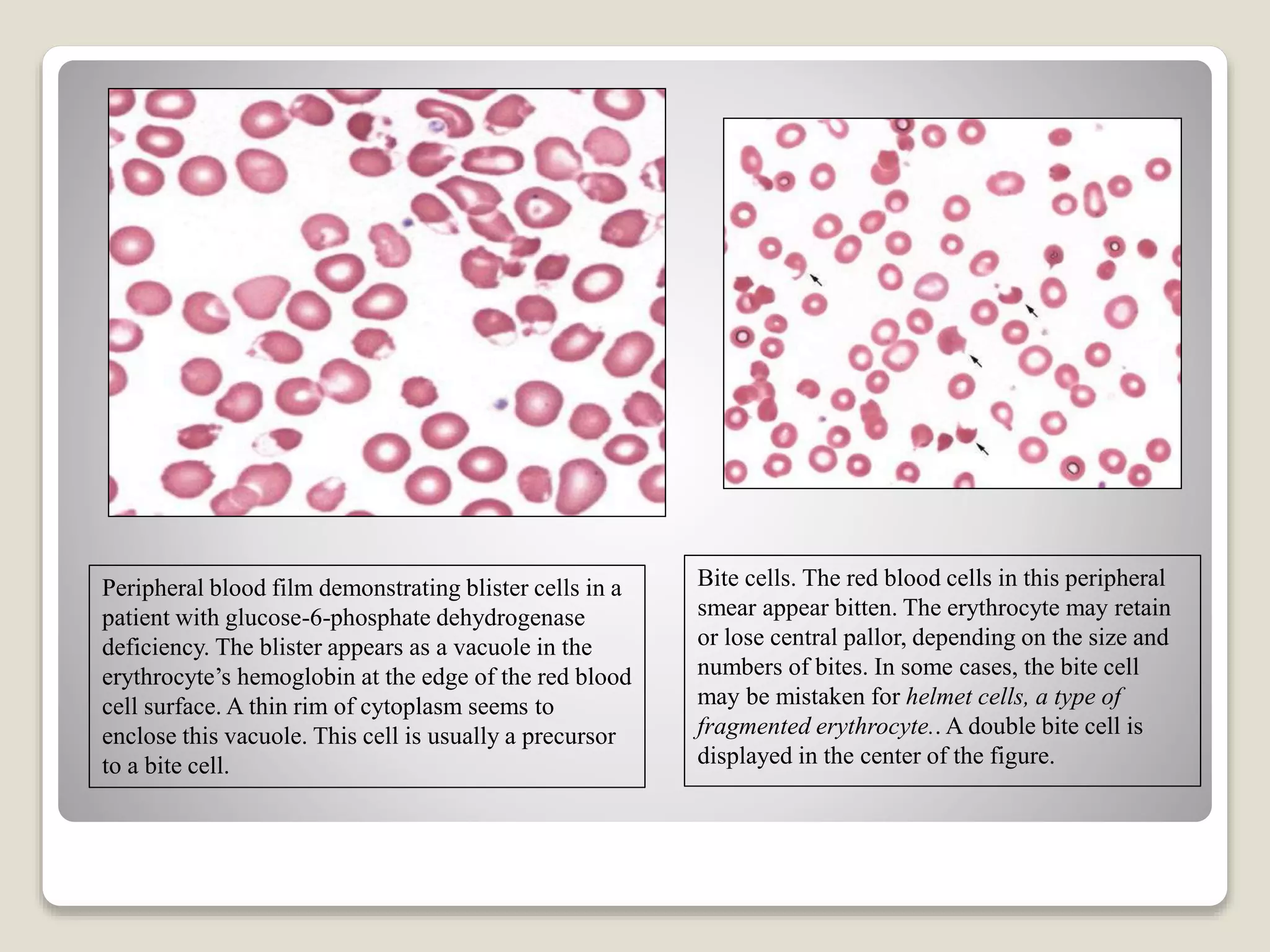
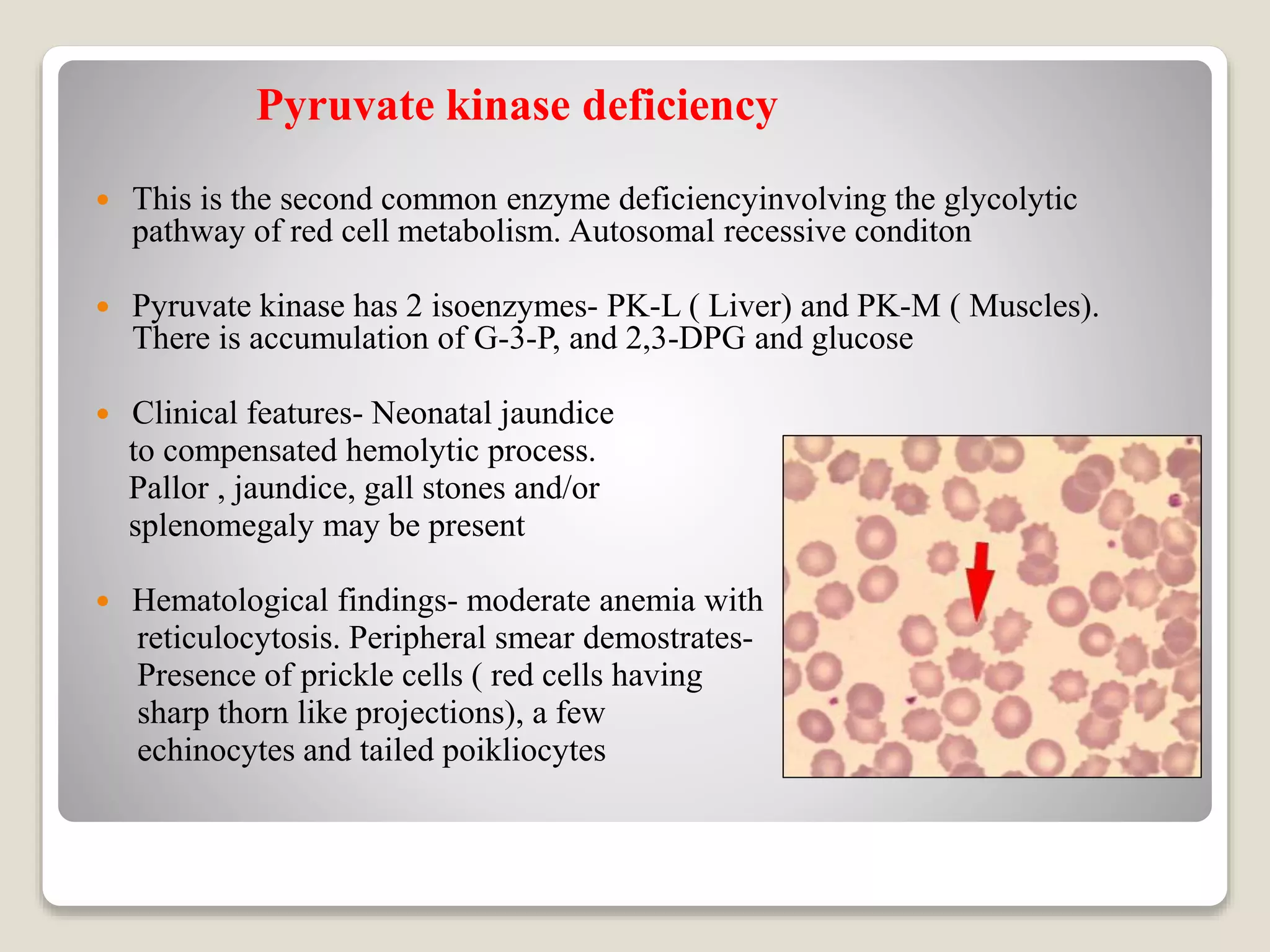
This document provides an overview of hemolytic anemia, including definitions, pathogenesis, classification, clinical features, laboratory findings, and approaches. Hemolytic anemia is characterized by increased red blood cell destruction. It can be hereditary or acquired. Specific hereditary forms discussed include hereditary spherocytosis, elliptocytosis, and pyropoikilocytosis, which are caused by red blood cell membrane defects. Clinical features may include pallor, jaundice, splenomegaly, and gallstones. Laboratory findings aid in diagnosis and include peripheral smear showing abnormal red blood cells, reticulocytosis, and elevated bilirubin. The document also discusses hemolytic anemia evaluation and differential diagnoses.