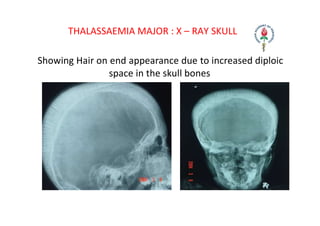
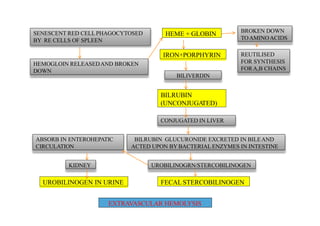
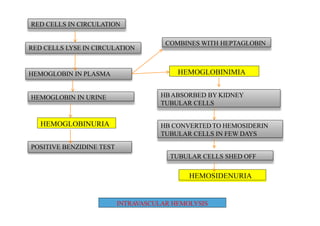
Hemolytic anemia results from increased red blood cell destruction coupled with the bone marrow's ability to increase red blood cell production in response. There are two types: congenital/hereditary forms caused by genetic defects, and acquired forms caused by external factors. Common hereditary types include thalassemia from hemoglobin defects, sickle cell anemia from abnormal hemoglobin, and G6PD deficiency from enzyme defects. Symptoms include anemia, jaundice, splenomegaly. Diagnosis involves blood tests showing markers of hemolysis like low haptoglobin, increased LDH and bilirubin. Treatment depends on the underlying cause but may include blood transfusions, iron chelation therapy,







![INTRACORPUSCULAR
DEFECTS (INTRINSIC)
EXTRACORPUSCULAR
FACTORS (EXTRINSIC)
Hereditary • Hemoglobinopathies
• Enzymopathies
• Membrane
Cytoskeletal Defects
• Familial Hemolytic
Uremic Syndrome
Acquired • Paroxysmal
Nocturnal
Hemoglobinuria
• Mechanical
Destruction
[Microangiopathic]
• Toxic Agents
• Drugs
• Infectious
• Autoimmune](https://image.slidesharecdn.com/hemolytic-anemia-230427043550-77d3e330/85/Hemolytic-Anemia-pptx-10-320.jpg)
![GENERAL FEATURES OF HEMOLYTIC DISORDERS
General examination ‐ Jaundice, pallor ,bossing of
skull
Systemic examination findings ‐ Enlarged spleen
Hemoglobin
MCV
Reticulocytes
Bilirubin
LDH
‐ From normal to severely reduced
‐ usually increased
‐ increased
‐ increased[mostly unconjugated]
‐ increased](https://image.slidesharecdn.com/hemolytic-anemia-230427043550-77d3e330/85/Hemolytic-Anemia-pptx-11-320.jpg)