Downloaded 483 times








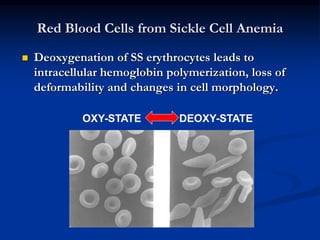



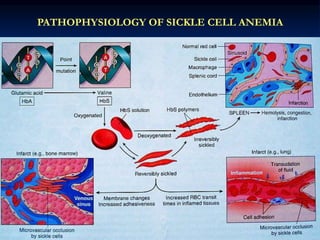
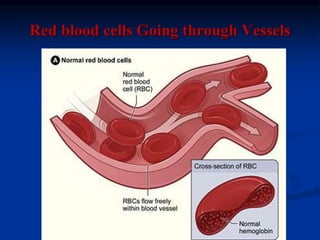
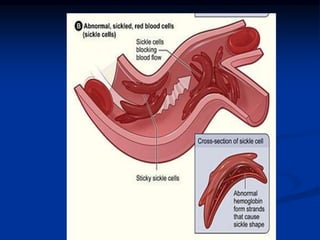






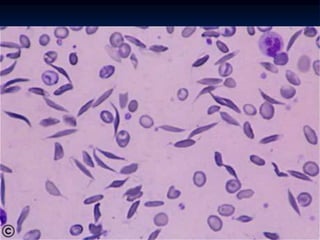
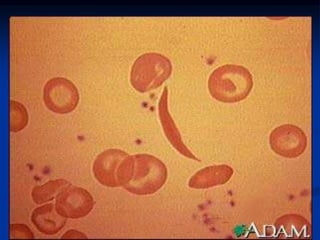
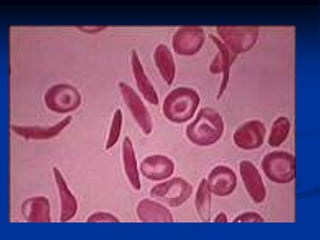
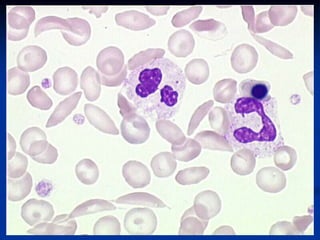


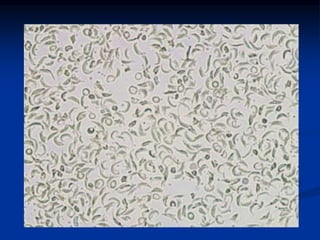




Sickle cell anemia is caused by a genetic mutation that results in abnormal hemoglobin called HbS. When HbS is deoxygenated, it polymerizes inside red blood cells, causing them to take on a sickle or holly leaf shape. This leads to hemolysis, anemia, vaso-occlusive crises involving painful blockages in small blood vessels, and organ damage. The condition is most common where malaria is endemic, as the sickle cell trait provides resistance to that disease. Laboratory testing can demonstrate sickle cells on blood smears and the presence of HbS on electrophoresis.











































































































