Downloaded 538 times










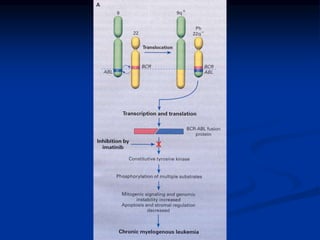
![Pathogenesis
Genetic abnormality
CML is the result of an acquired genetic abnormality
A translocation between chromosome 9 and 22 [t(9;22)] –
the Philadelphia chromosome
The oncogene BCR-ABL encodes an enzyme – tyrosine
phosphokinase (usually p210)](https://image.slidesharecdn.com/chronicleukemias-160524143702/85/Chronic-leukemias-10-320.jpg)











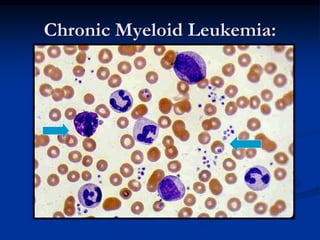
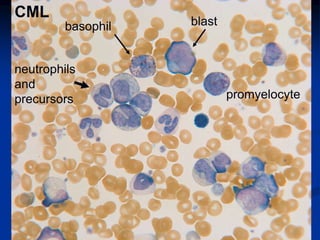
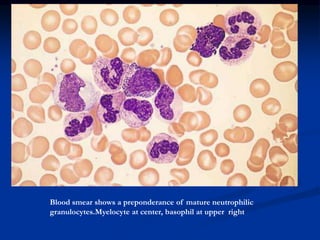
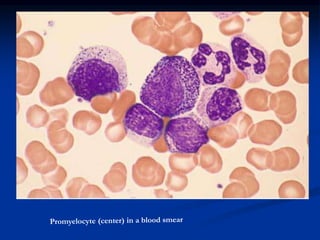
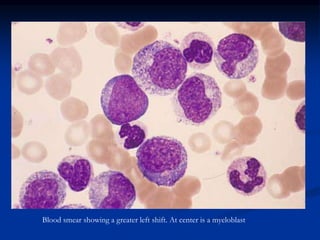
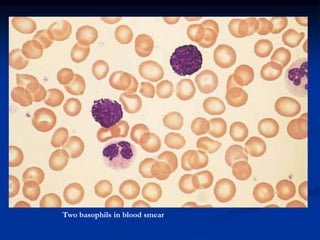
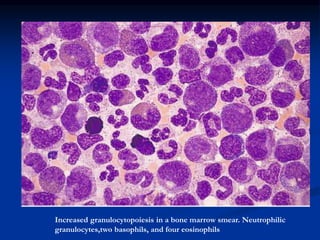







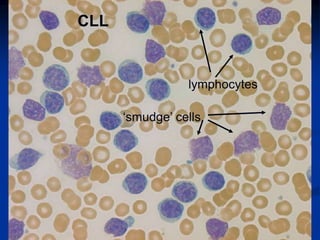
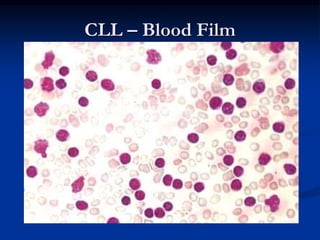
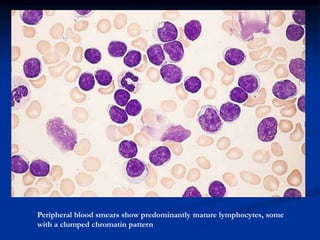
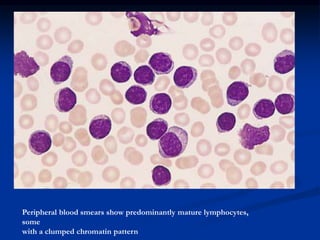
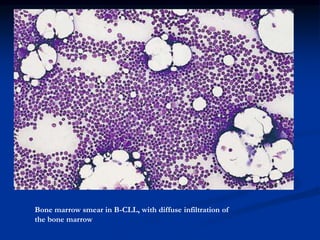




Chronic leukemias have an insidious onset and are usually less aggressive than acute leukemias. The two main types are chronic myeloid leukemia, characterized by the Philadelphia chromosome, and chronic lymphocytic leukemia, which mainly affects B cells. These diseases involve increased numbers of mature but dysfunctional white blood cells and are diagnosed based on blood counts, bone marrow examination, and identification of genetic abnormalities.



































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)