Downloaded 634 times

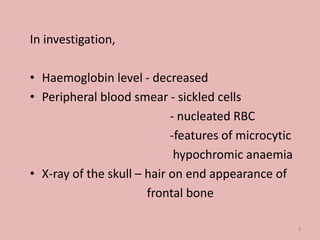


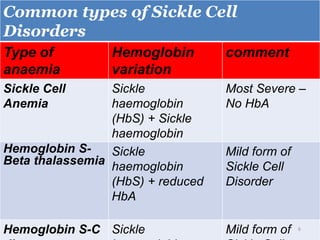
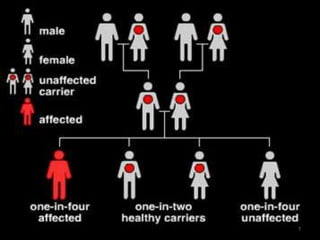



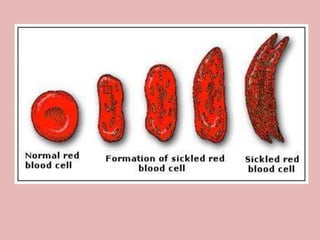
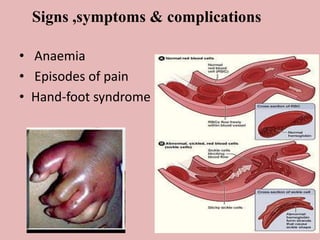

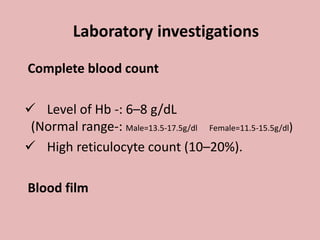
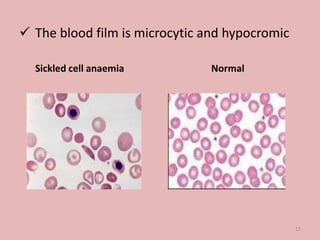
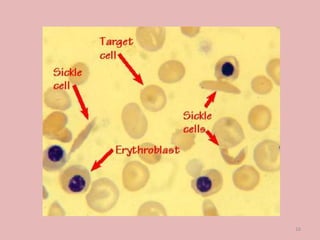

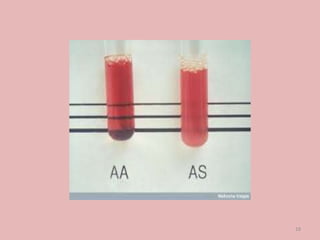

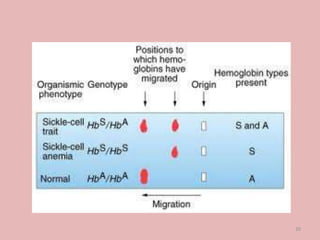
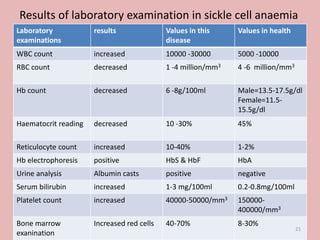
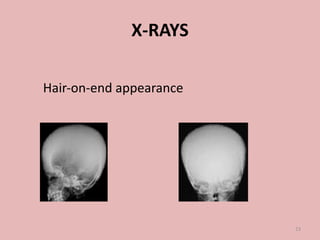
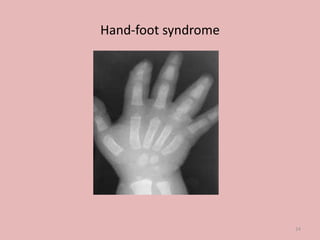
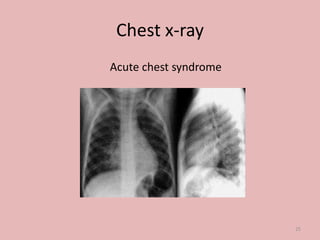
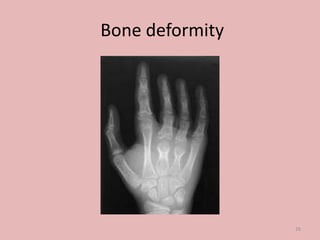
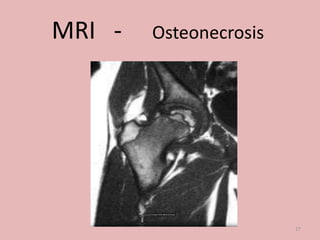




A 4-year-old boy presented with severe back pain and vomiting. Examination found pallor and spleen enlargement. Tests showed decreased hemoglobin, sickled red blood cells, and hair-on-end skull appearance consistent with sickle cell anemia. Sickle cell anemia is an inherited blood disorder caused by a mutation causing hemoglobin to sickle and break down red blood cells early, resulting in anemia. Common complications include pain crises, infections, and organ damage. Diagnosis is confirmed by hemoglobin electrophoresis showing elevated hemoglobin S.



































































