Downloaded 565 times



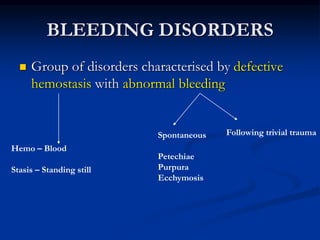
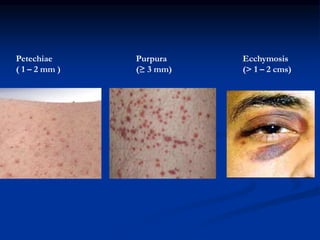



















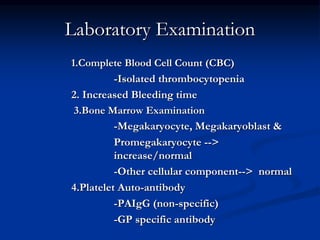
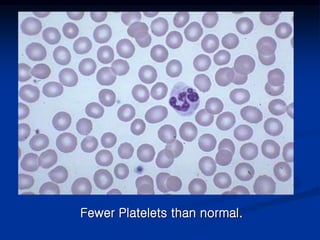
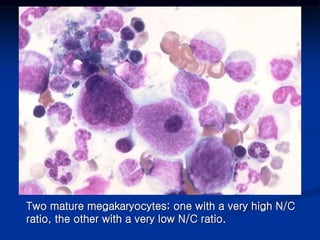
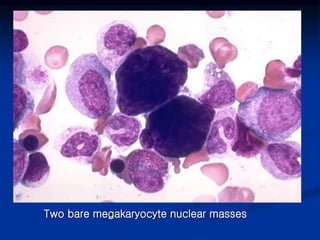



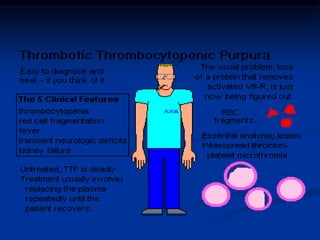
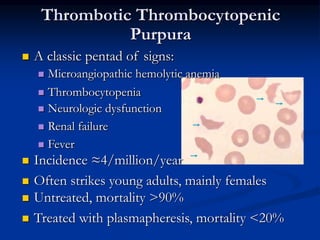





Thrombocytopenia can be classified as quantitative (reduced platelet count) or qualitative (platelet function defects). Idiopathic thrombocytopenic purpura (ITP) is an immune-mediated disorder causing reduced platelet counts. It typically presents with purpura and petechiae. Diagnosis involves isolated thrombocytopenia with normal or increased megakaryocytes on bone marrow exam and platelet autoantibodies. Treatment involves corticosteroids, splenectomy, or IVIG. Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy with a classic pentad of signs: microangiopathic hemolytic anemia, thromb















































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)






![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
