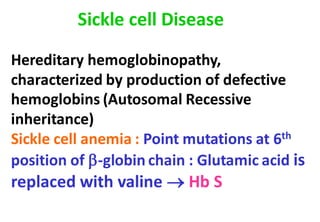
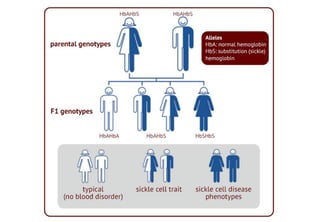
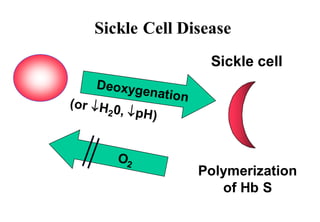
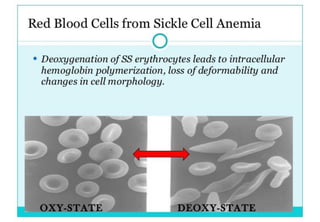
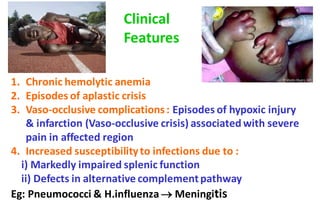
This document discusses sickle cell anemia, beginning with a description of normal hemoglobins and hemoglobinopathies. It then focuses on sickle cell anemia, caused by a point mutation replacing glutamic acid with valine in the beta globin chain, resulting in hemoglobin S (HbS). Homozygous individuals have >80% HbS and experience the full manifestations of sickle cell anemia. Clinical features include chronic hemolytic anemia, episodes of aplastic crisis and vaso-occlusive complications resulting in severe pain, as well as increased susceptibility to infections due to impaired splenic function and complement pathway defects.