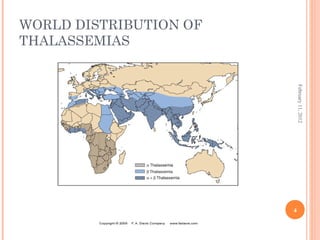
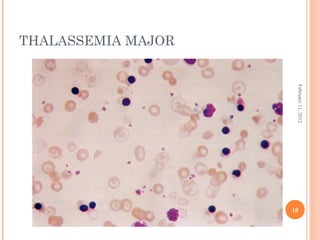
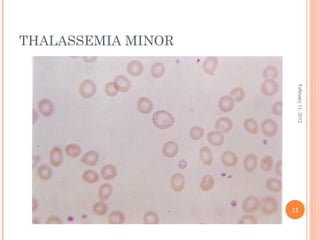
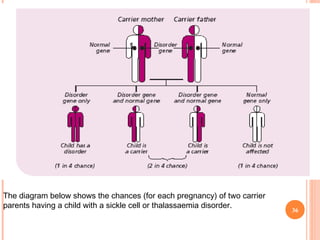
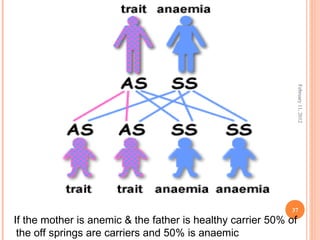
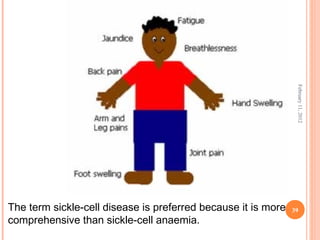
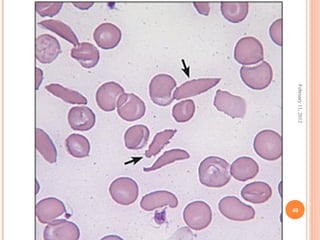
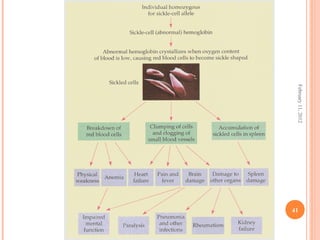
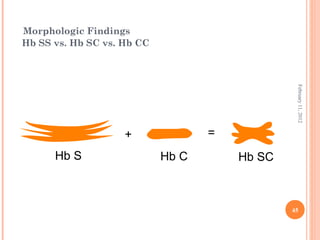
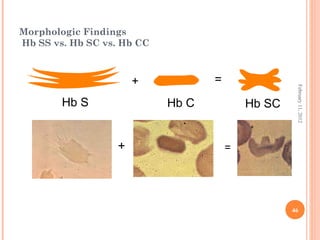
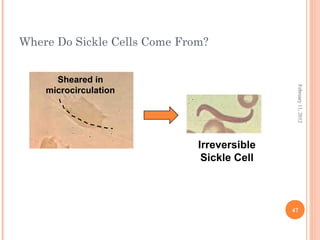
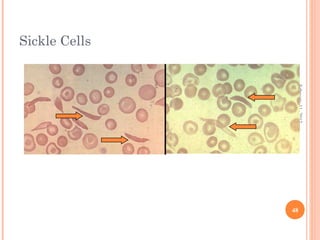
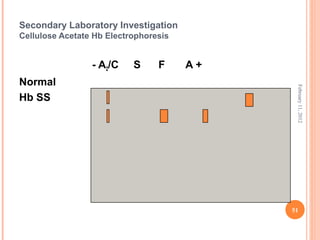
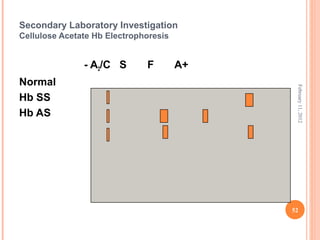
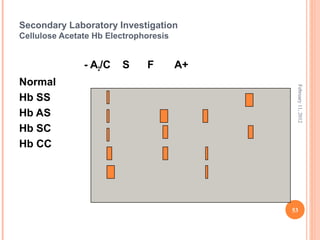
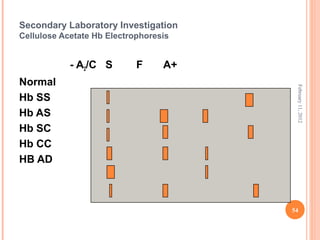
Dr. Neela Ferdoushi presented on thalassemias and hemoglobinopathies. Thalassemias are genetic disorders caused by mutations that decrease the synthesis of the alpha or beta globin chains, resulting in imbalanced globin chain production and anemia. The clinical severity depends on the specific gene mutations inherited. Untreated homozygous forms can be fatal in childhood. Heterozygous carriers may be asymptomatic or have mild anemia. Laboratory tests can help identify abnormalities in hemoglobin type or amounts through hemograms and hemoglobin electrophoresis. Management involves blood transfusions and iron chelation therapy for severe forms.