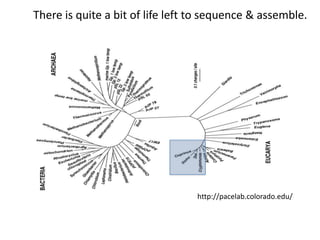
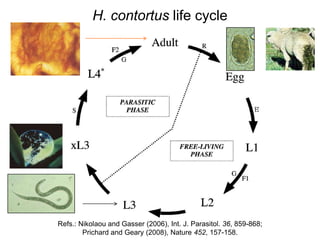
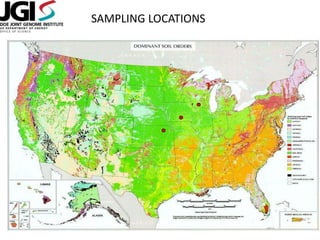
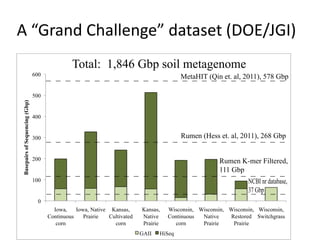
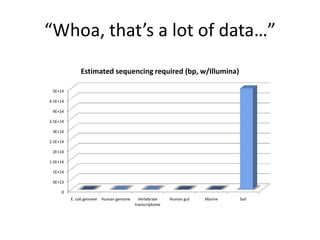
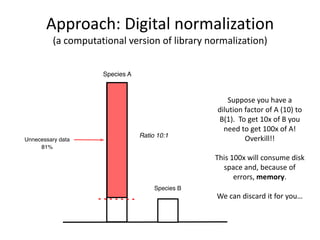
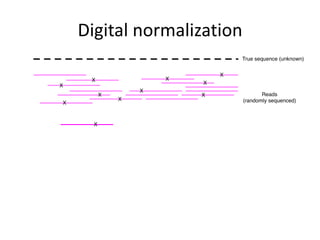
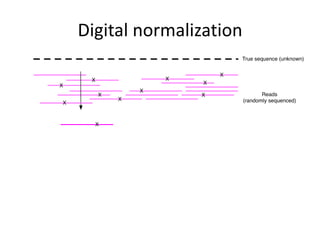
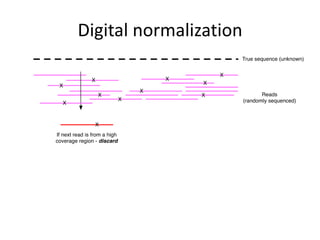
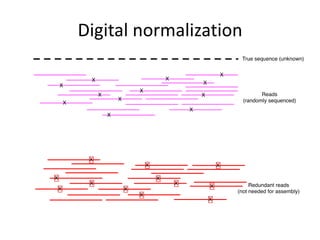
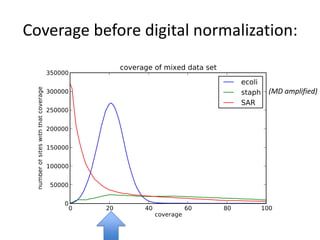
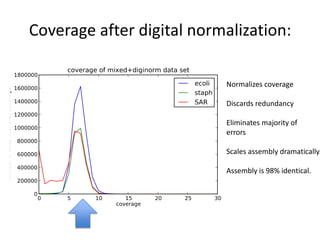
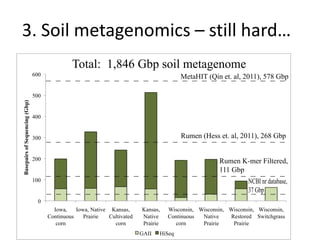
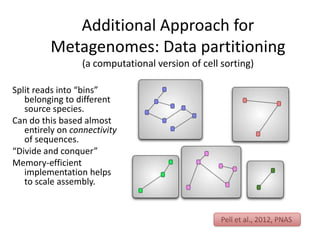
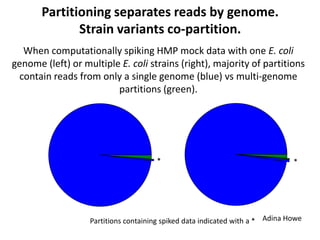
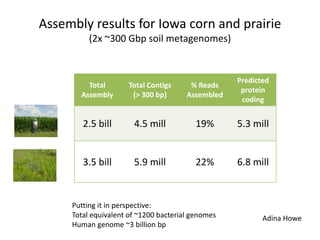
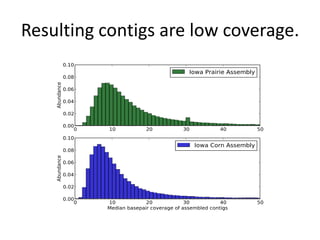
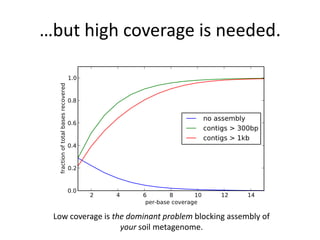
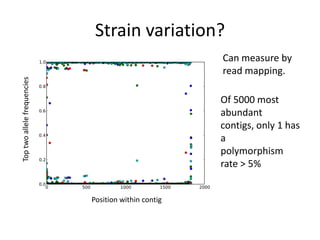
This document discusses challenges and approaches for assembling large metagenomic and genomic datasets using short read sequencing data. Three main challenges are discussed: 1) Assembling the parasitic nematode H. contortus genome due to high polymorphism and repeats. Digital normalization helped enable assembly by reducing redundancy and errors. 2) Assembling the lamprey transcriptome with no reference and too much data. Digital normalization reduced the data volume and enabled assembly. 3) Assembling large soil metagenomic datasets, which are difficult due to their scale and complexity. Data partitioning separates reads into bins to enable "divide and conquer" assembly approaches. While progress has been made, challenges around strain variation, scaffolding, and connecting














































![[2013.10.29] albertsen genomics metagenomics](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131029070115-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)









































































