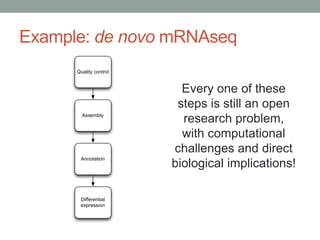
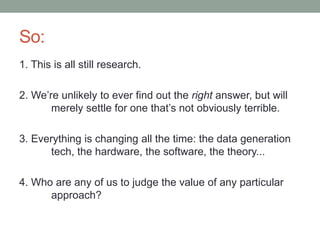
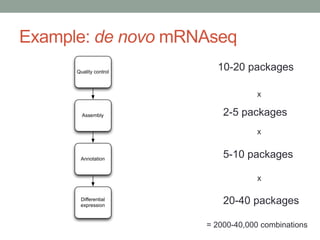
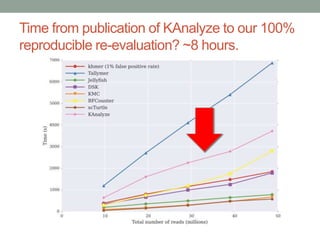
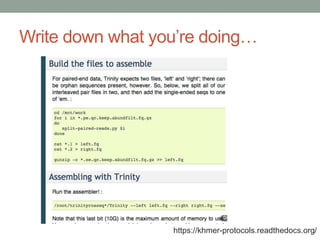
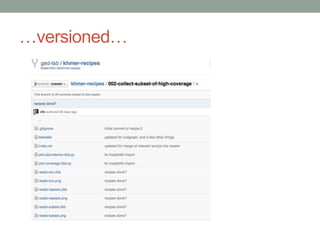
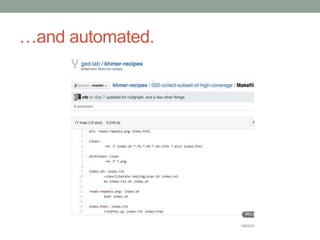
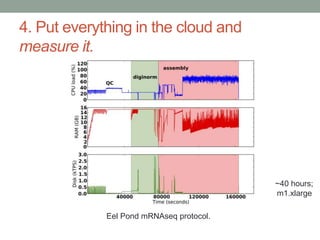
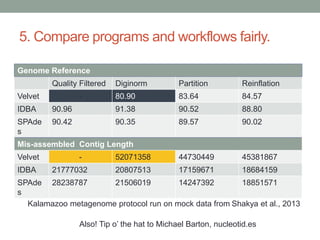
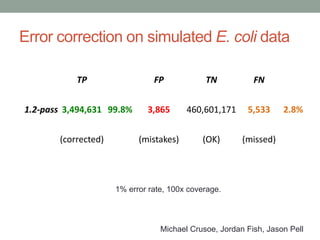
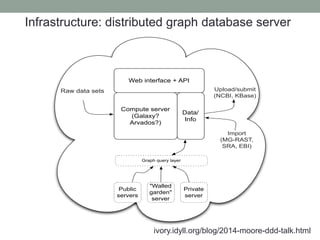
This document summarizes a keynote presentation about challenges in bioinformatics software development and proposed solutions. Some of the key points made include: 1) bioinformatics software development involves multiple disciplines including computer science, software engineering, statistics, and biology, each with different priorities; 2) there is a massive proliferation of bioinformatics software packages that leads to many difficult choices for researchers; 3) proposed solutions include developing software in a more modular and automated way, using common benchmarks and protocols to evaluate tools, and focusing on reproducibility and usability.



![So I said these things…
“this tipping point was exacerbated by the loss of about
80% of the worlds data scientists in the 2021 Great
California Disruption.”
“[ Benchmarks ] have proven to be stifling of innovation,
because of the tendency to do incremental improvement.”
ivory.idyll.org/blog/2014-bosc-keynote.html](https://image.slidesharecdn.com/2014-abic-talk-141011171747-conversion-gate02/85/2014-abic-talk-5-320.jpg)
![So I said these things…
“this tipping point was exacerbated by the loss of about
80% of the worlds data scientists in the 2021 Great
California Disruption.”
“[ Benchmarks ] have proven to be stifling of
innovation, because of the tendency to do incremental
improvement.”
ivory.idyll.org/blog/2014-bosc-keynote.html](https://image.slidesharecdn.com/2014-abic-talk-141011171747-conversion-gate02/85/2014-abic-talk-6-320.jpg)