




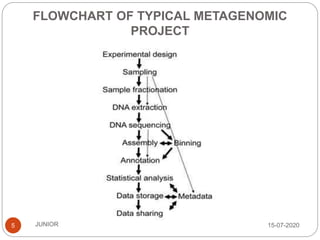



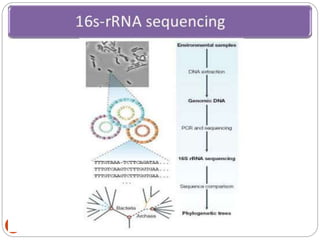








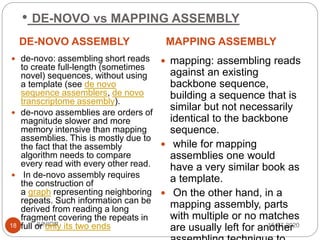






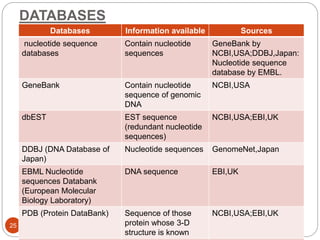

















Metagenomics, a term first introduced in 1998, refers to analyzing environmental genes in a manner akin to studying single genomes, shedding light on diverse microbial communities. Early studies primarily focused on 16S ribosomal RNA sequences, which helped uncover numerous uncultured organisms, while modern approaches have expanded due to advancements in sequencing technologies and the identification of new viral and bacterial species in various environments. The field has significant applications in agriculture, biofuels, biotechnology, ecology, environmental remediation, and infectious disease diagnostics.
















































































