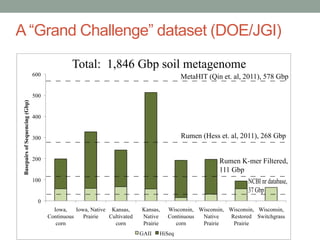
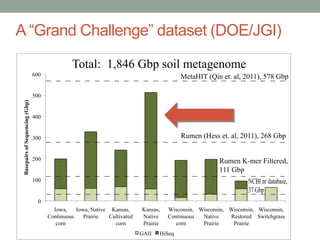
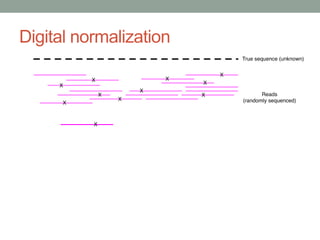
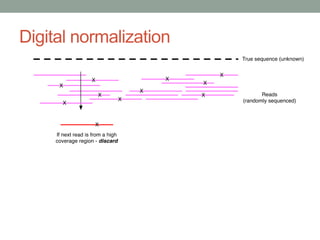
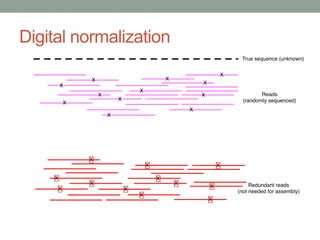
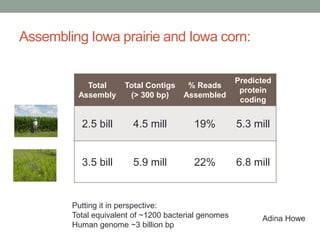
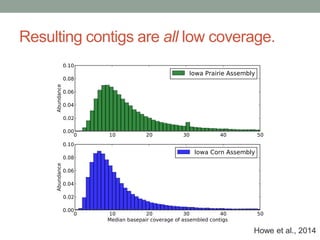
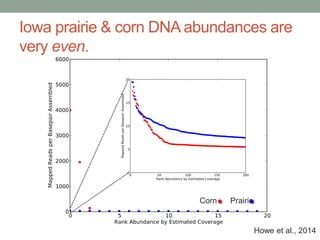
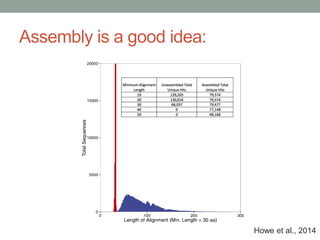
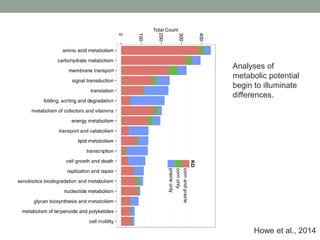
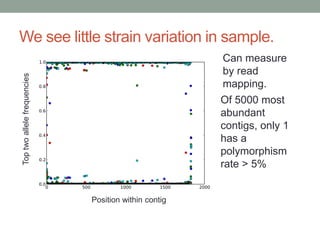
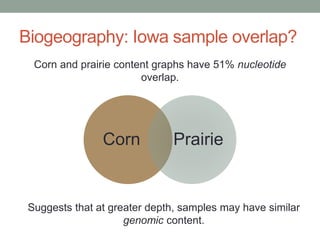
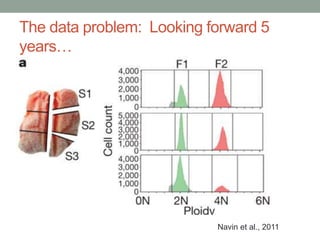
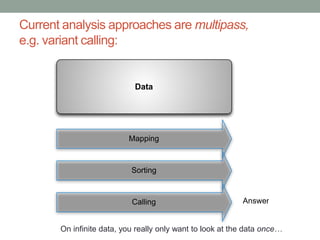
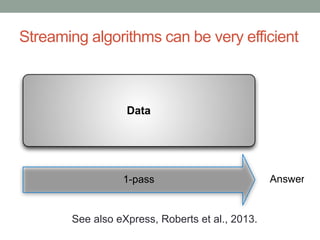
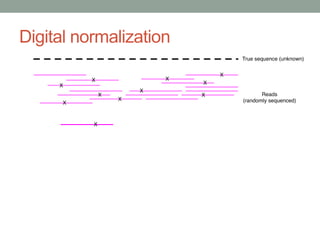
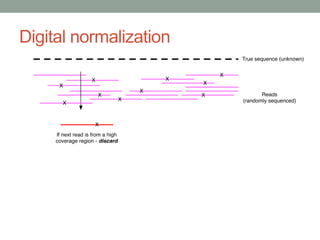
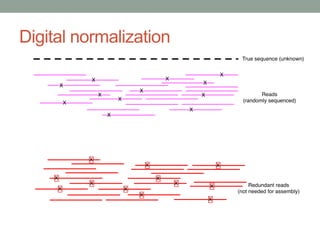
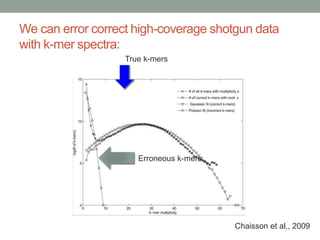
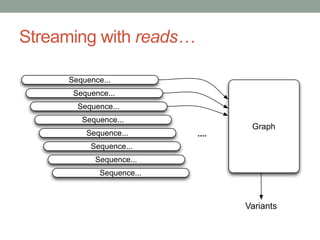
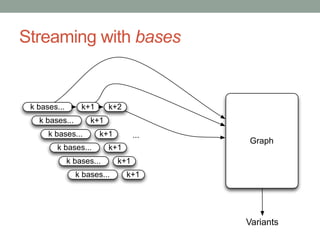
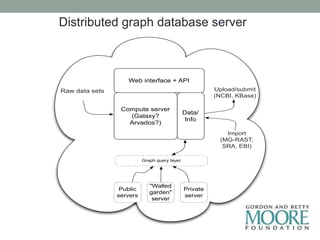
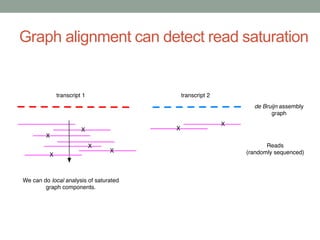
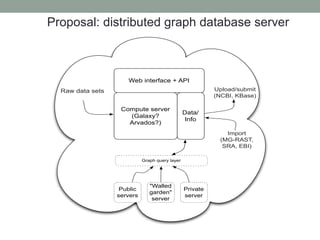
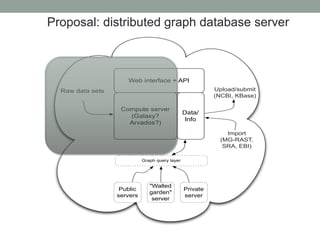
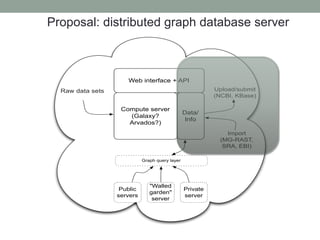
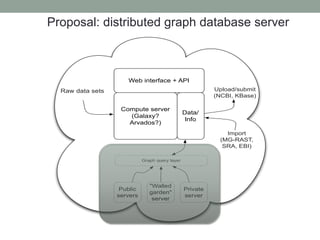
This document discusses scalable computational approaches for exploring microbial diversity using metagenomic sequencing data. It describes a "digital normalization" algorithm that uses a streaming computational approach to lossy compression of sequencing data in a memory- and time-efficient way. This allows assembly and analysis of very large soil metagenomic datasets totaling over 1.8 terabases. Comparison of Iowa prairie and corn field samples showed 51% nucleotide overlap, suggesting similar genomic content between these environments.












































































![[Infographic Korea Edition] The CEO Reputation Premium - Weber Shandwick](https://cdn.slidesharecdn.com/ss_thumbnails/ceo-ceo-150305194521-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)




























































