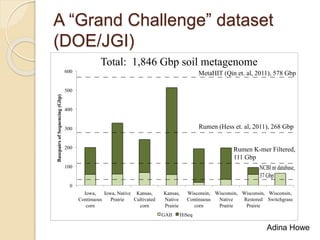
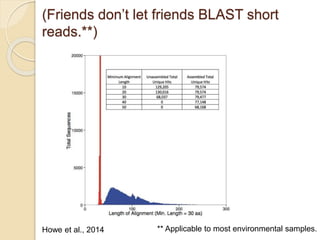
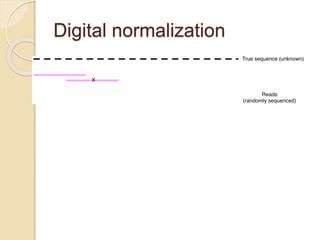
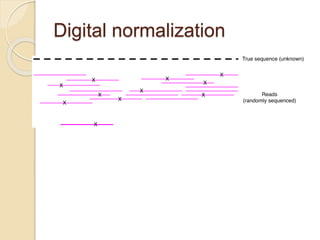
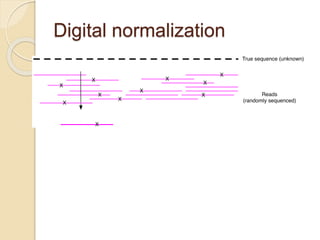
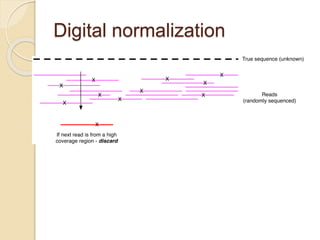
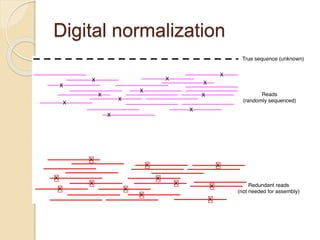
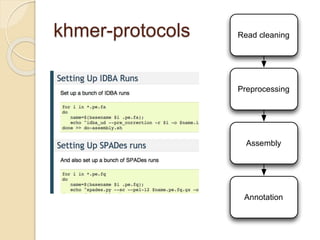
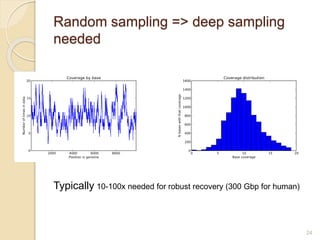
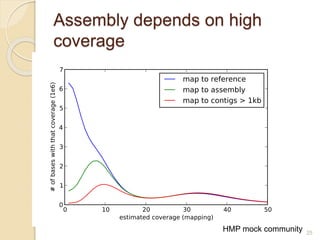
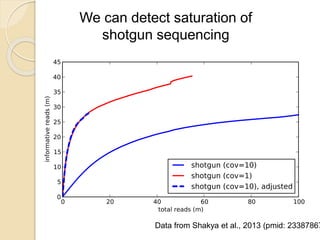
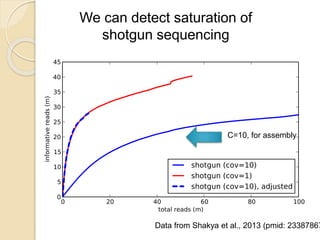
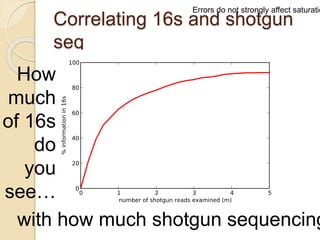
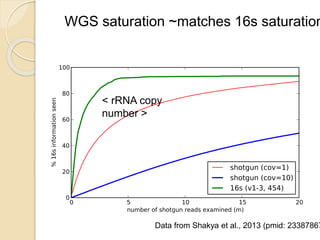
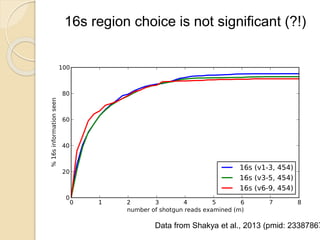
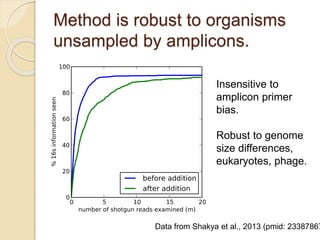
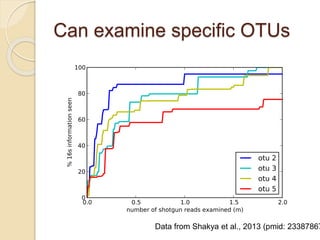
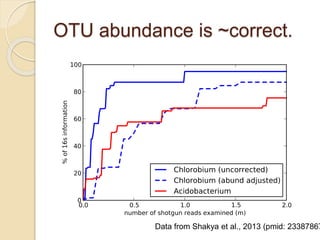
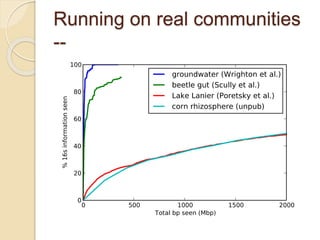
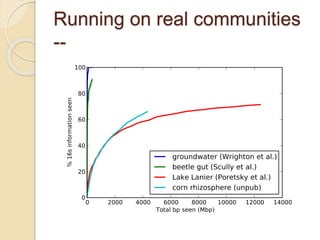
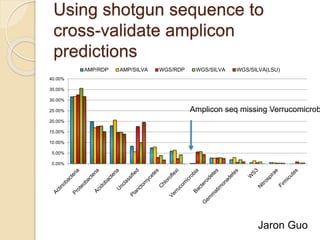
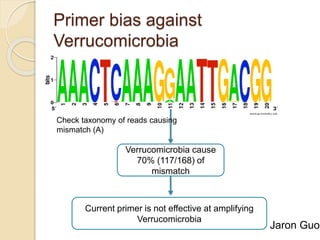
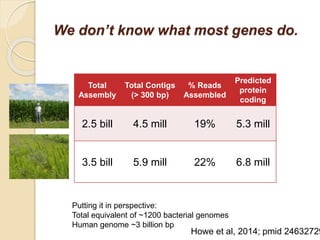
This document discusses assembling metagenomic sequences from soil samples to answer ecological questions. It describes a "Grand Challenge" dataset from the DOE/JGI of 1.8 Gbp of soil metagenome sequences. The author developed new computational methods called "cell sorting" and "library normalization" to assemble these large, diverse metagenomes into contigs over 2.5 Gbp in size. The author argues that assembly is now a largely solved problem and discusses using assembled sequences to annotate genes, reconstruct genomes and operons, study strain variation, and analyze phages and viruses. The document also explores using 16S amplicon sequencing to predict the depth of shotgun metagenomic sequencing needed and