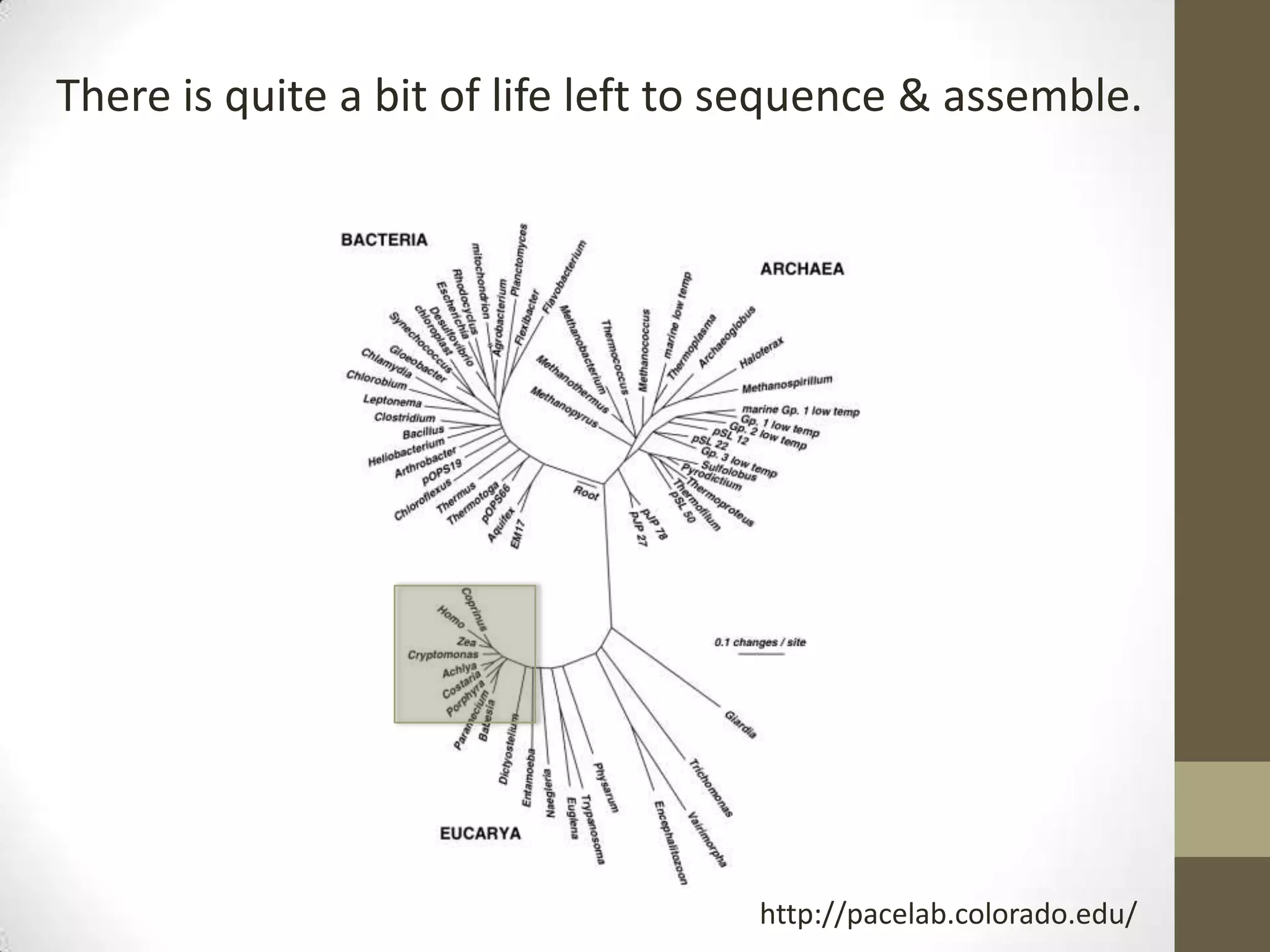
This document outlines Dr. Brown's research interests which focus on developing computational tools and approaches to analyze large and complex biological data sets from non-model organisms. Some key points include:
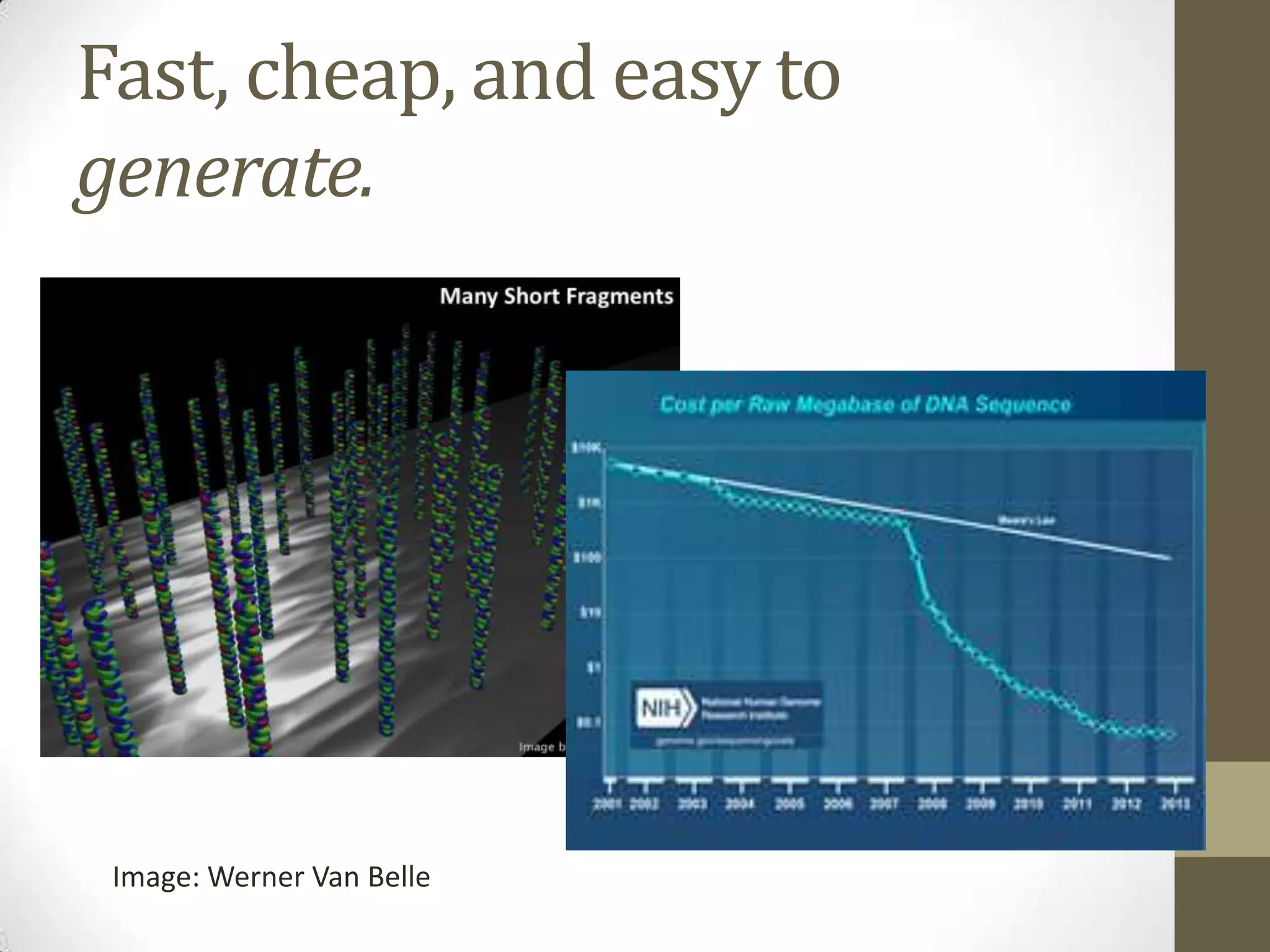
- His lab works on improving genome and transcriptome assembly from samples that are difficult to sequence such as metagenomes, single cells, and samples with high polymorphism.
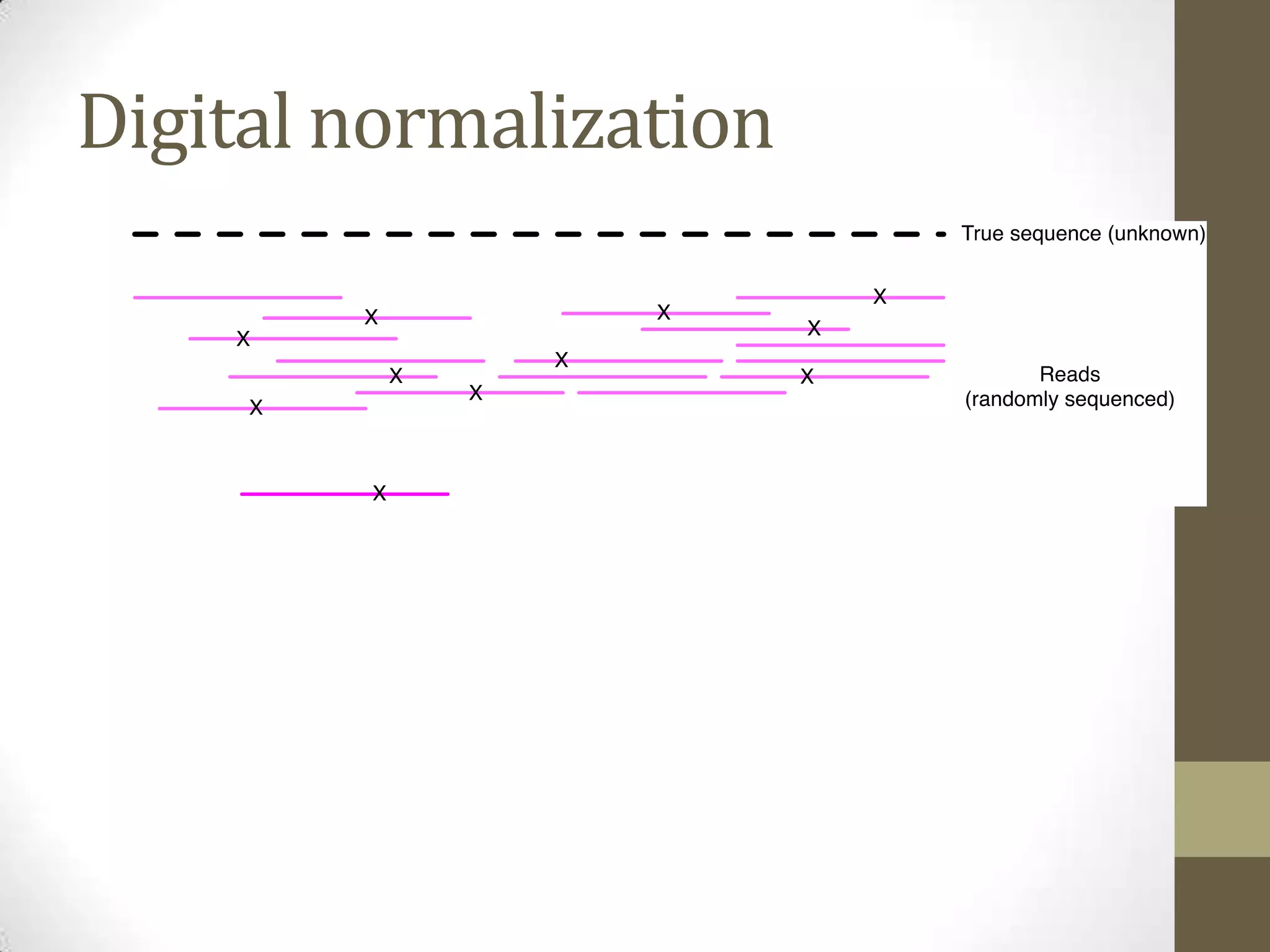
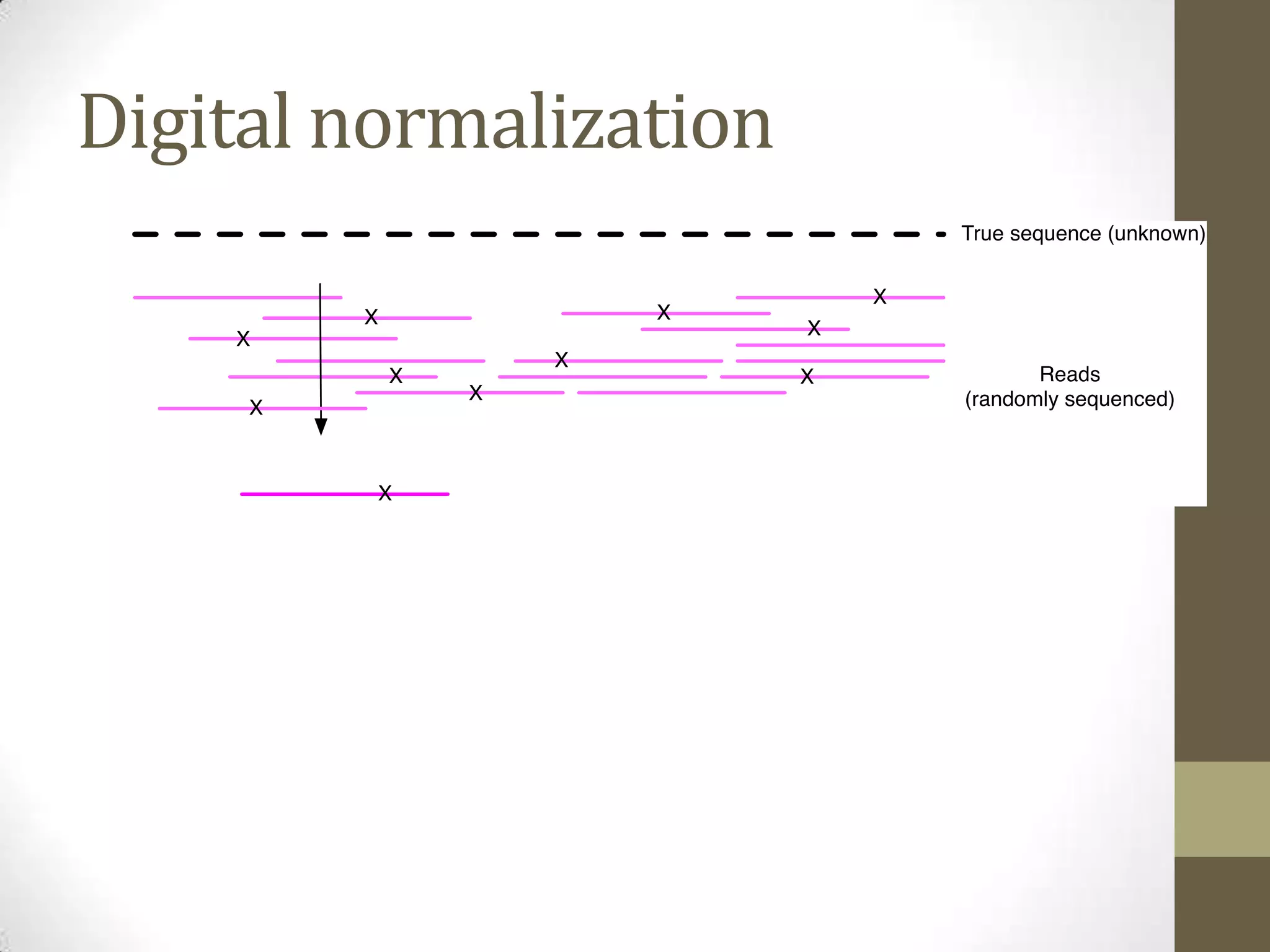
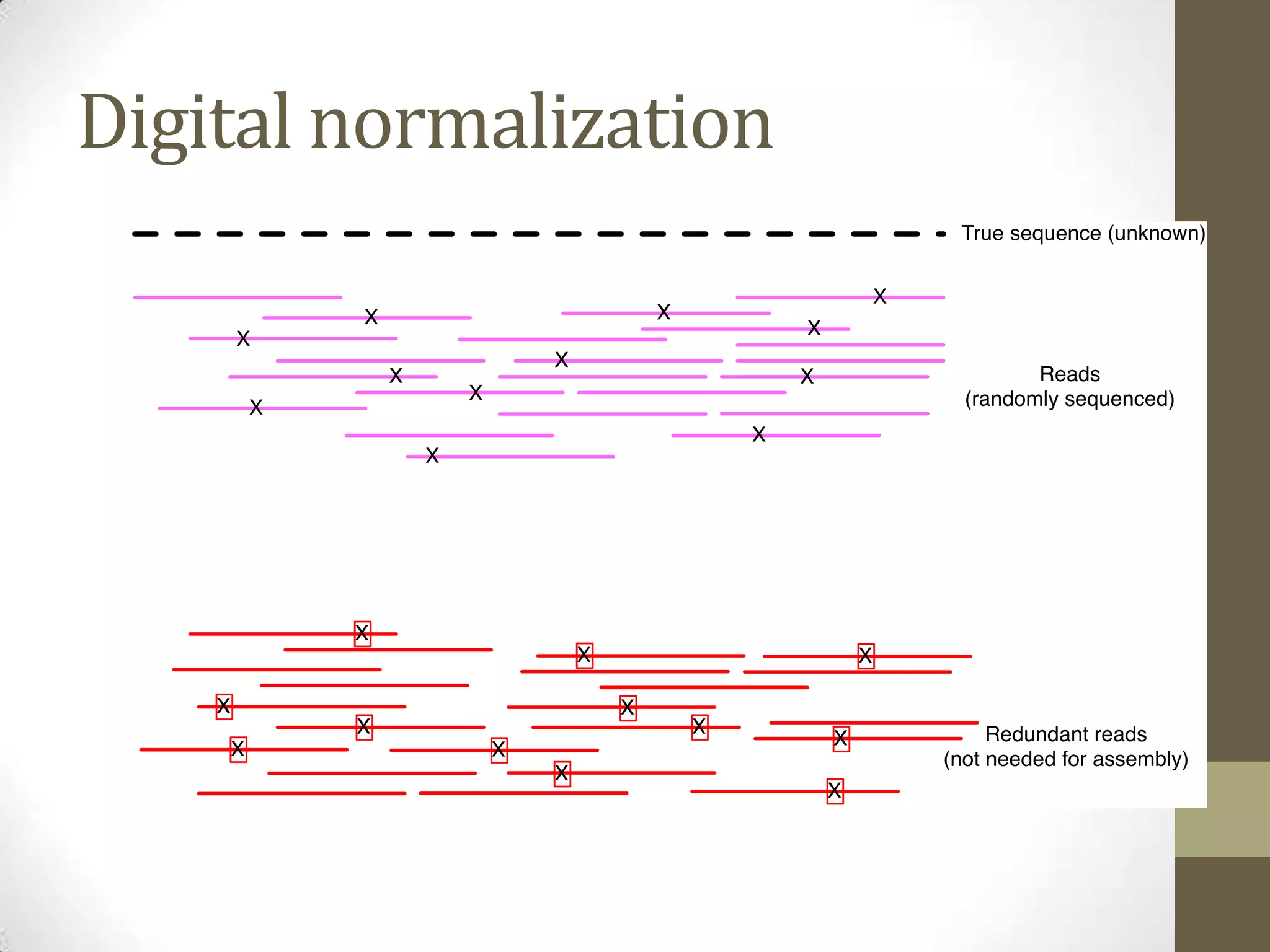
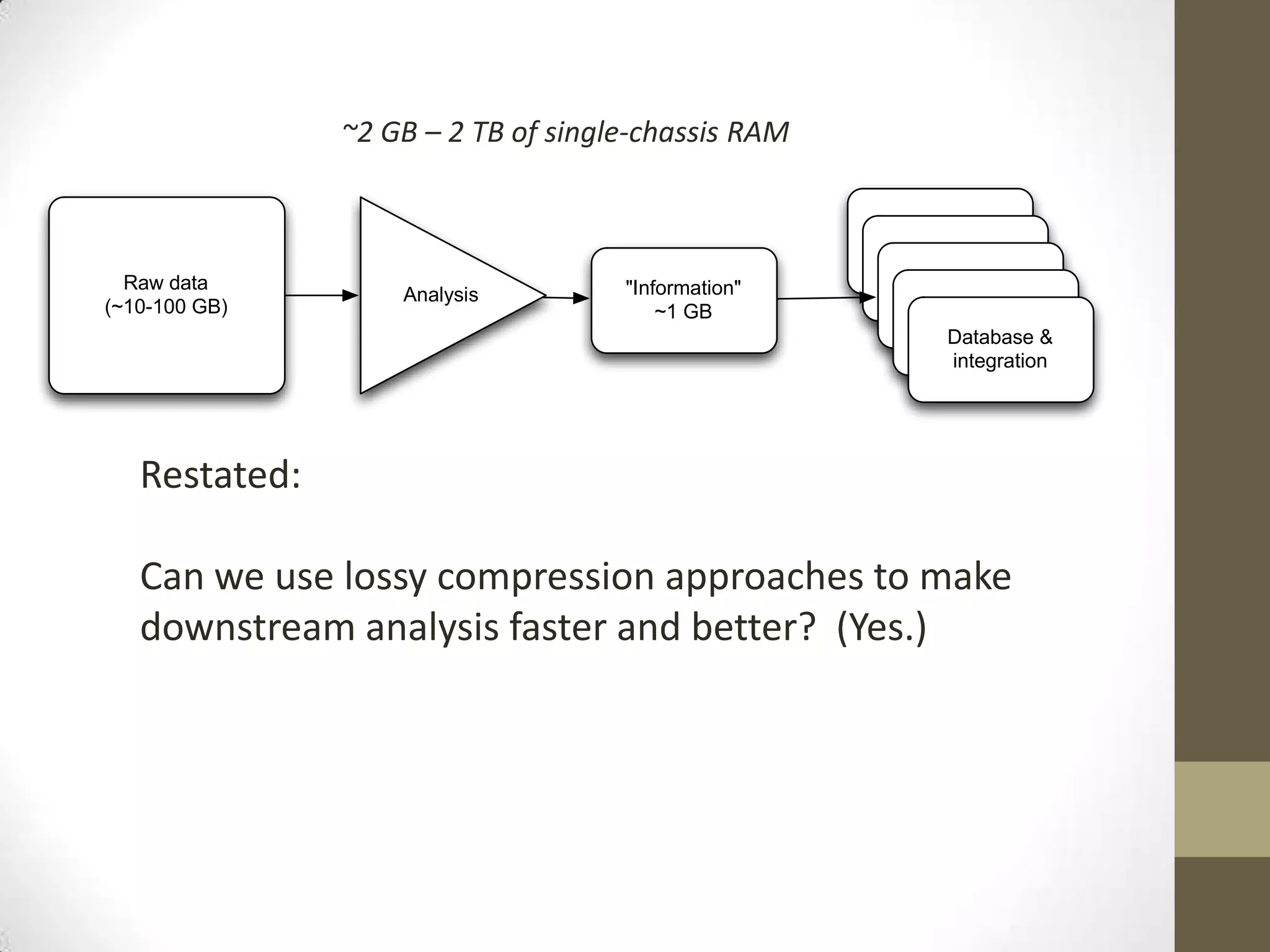
- He describes developing techniques like "digital normalization" which can speed up downstream data analysis by compressing raw data in a lossy but information-preserving way.
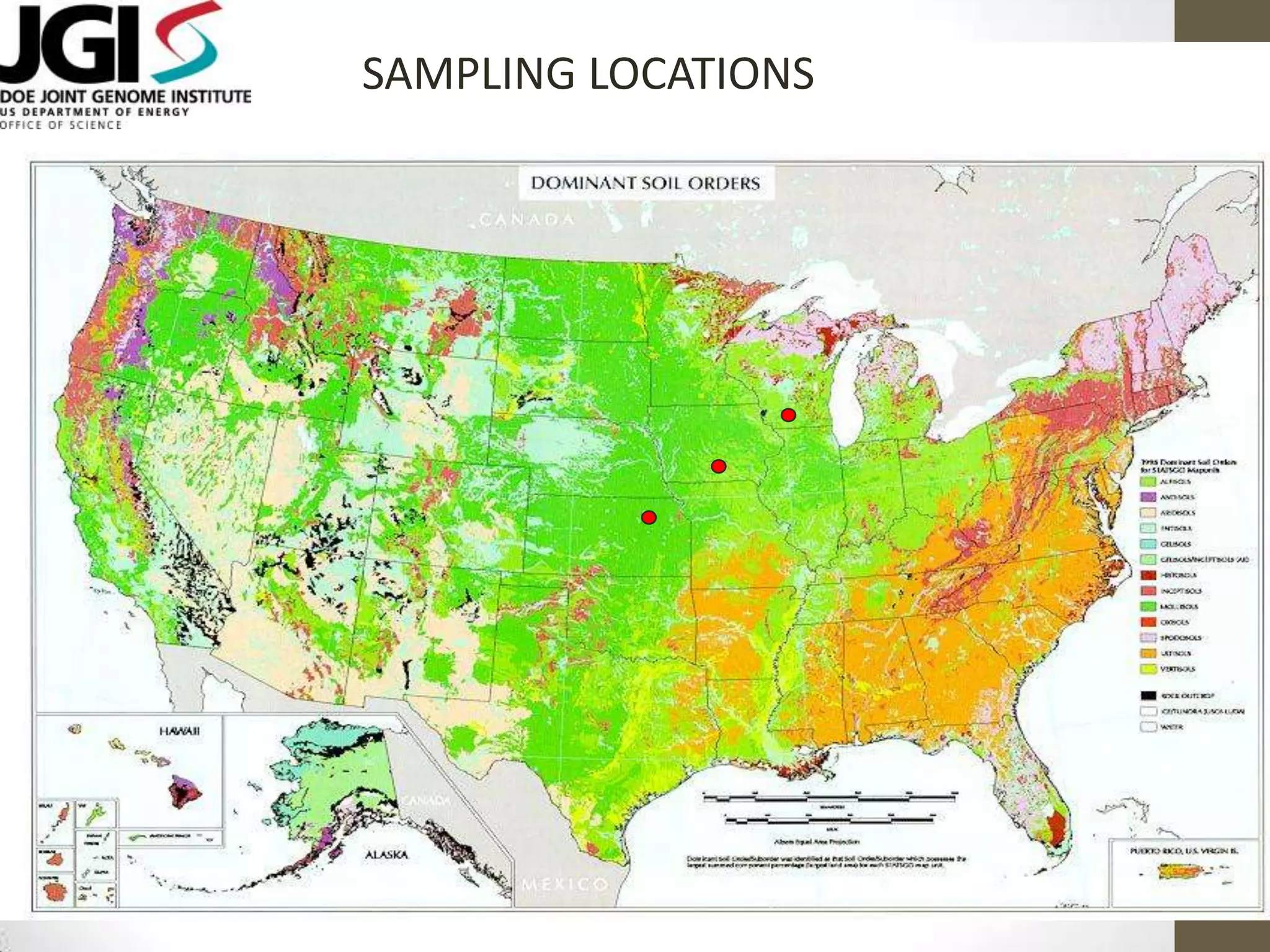
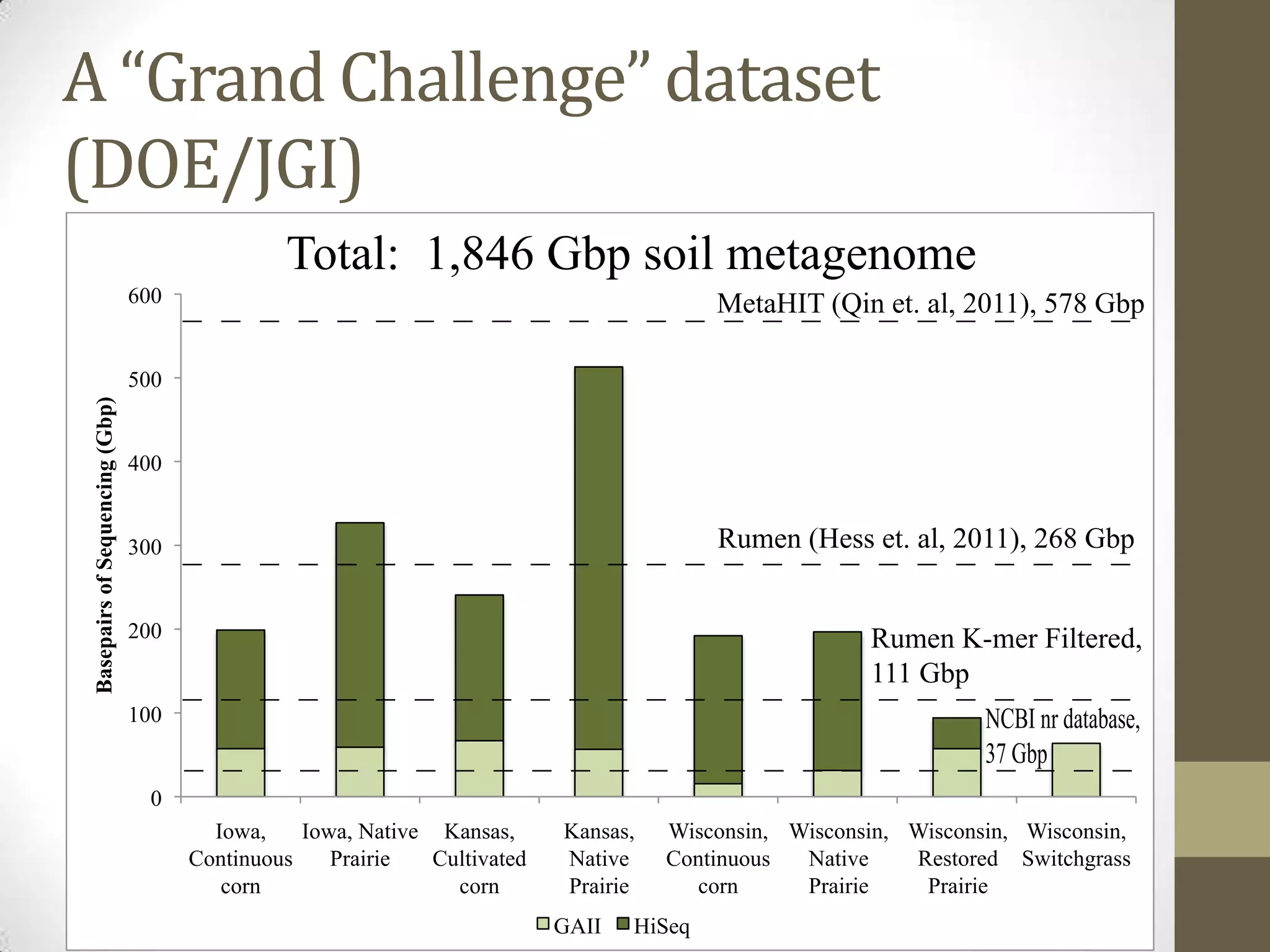
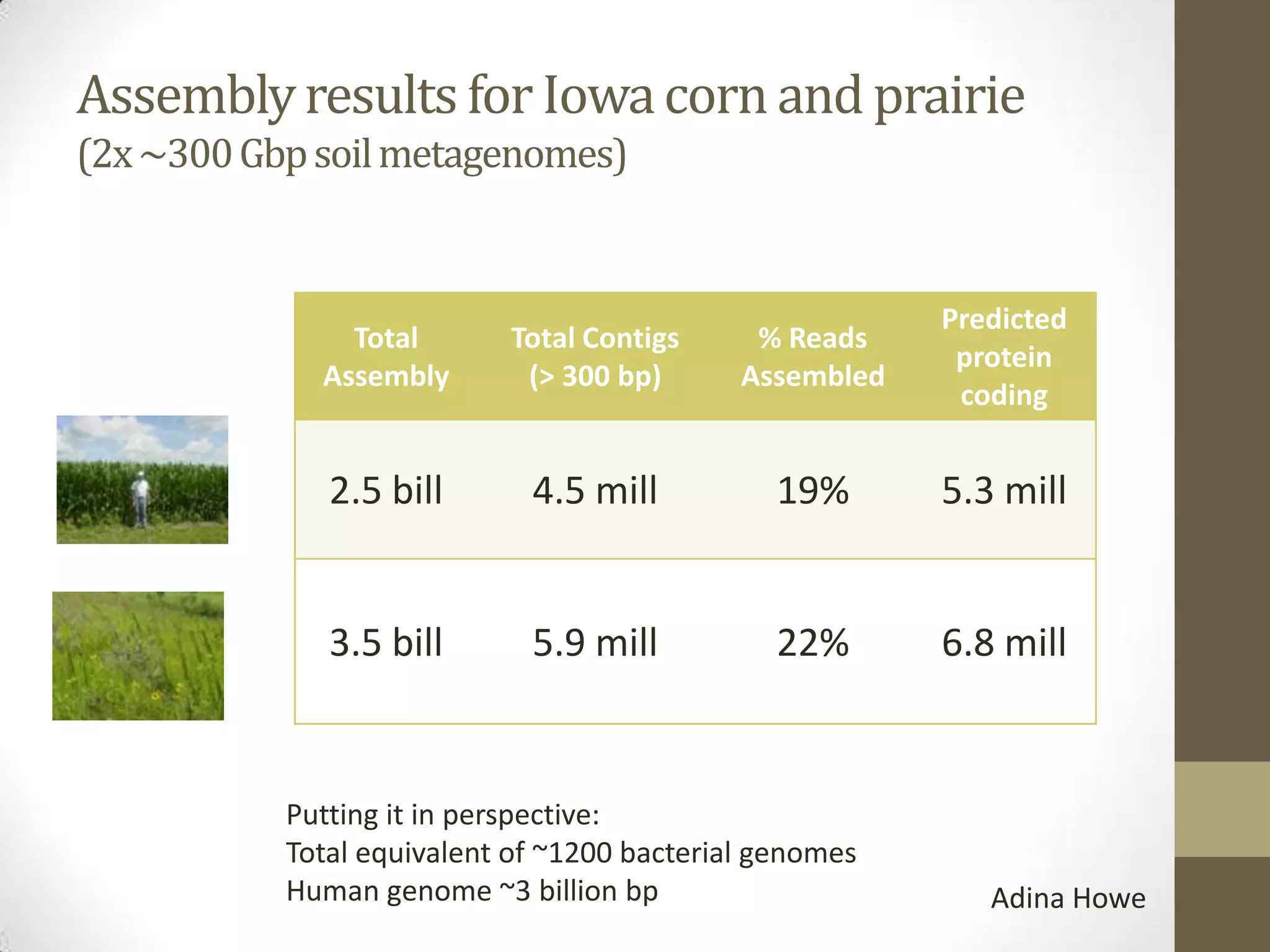
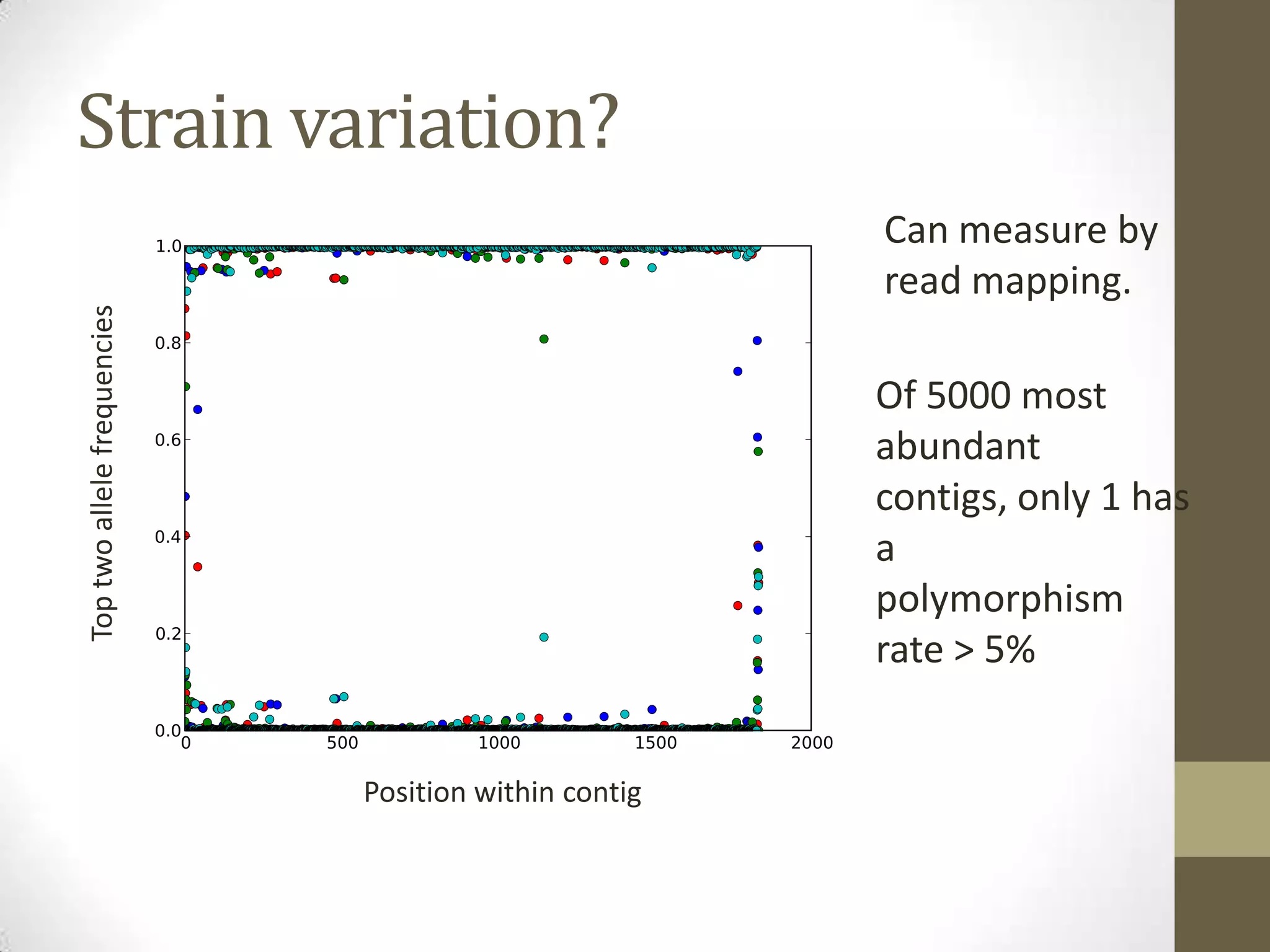
- An example project involved assembling 1.8 terabases of soil metagenome sequence data from various locations, finding little strain-level variation but that phage sequences were abundant










































![[TDC 2013] Integre um grid de dados em memória na sua Arquitetura](https://cdn.slidesharecdn.com/ss_thumbnails/tdc2013integrandocoherencearqttv1-130526184010-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




















































![[BDD 2025 - Mobile Development] Mobile Engineer and Software Engineer: Are we...](https://cdn.slidesharecdn.com/ss_thumbnails/md-mobileengineerandsoftwareengineerarewestillrelevantsidiqpermana-251127010650-55224ef1-thumbnail.jpg?width=640&height=640&fit=bounds)


![[BDD 2025 - Artificial Intelligence] Building AI Systems That Users (and Comp...](https://cdn.slidesharecdn.com/ss_thumbnails/ai-buildingaisystemsthatusersandcompanieslove-251124030845-038f7732-thumbnail.jpg?width=640&height=640&fit=bounds)







![[BDD 2025 - Full-Stack Development] PHP in AI Age: The Laravel Way. (Rizqy Hi...](https://cdn.slidesharecdn.com/ss_thumbnails/fs-phpinaiagethelaravelway-251125012602-ef9d330e-thumbnail.jpg?width=640&height=640&fit=bounds)




![[BDD 2025 - Full-Stack Development] The Modern Stack: Building Web & AI Appli...](https://cdn.slidesharecdn.com/ss_thumbnails/fs-themodernstackbuildingwebaiapplicationswithserverless-251124030844-388cf04f-thumbnail.jpg?width=640&height=640&fit=bounds)


