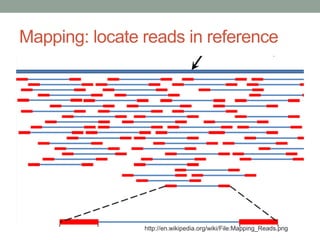
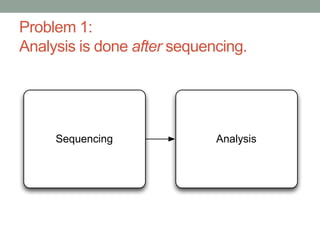
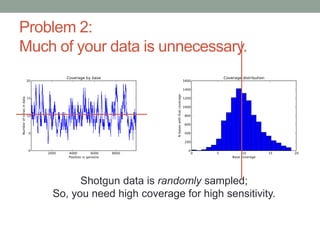
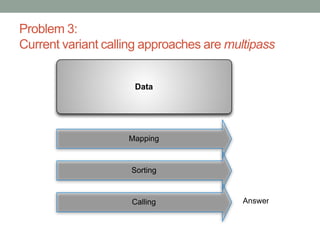
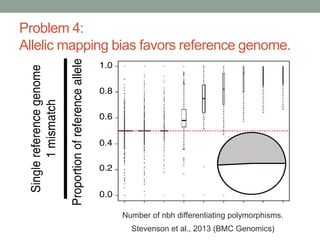
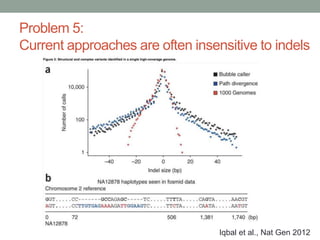
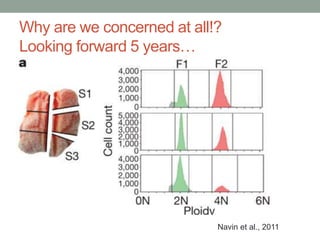
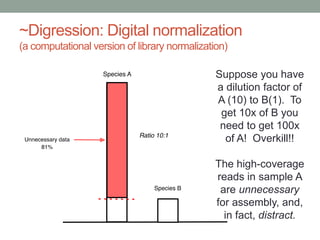
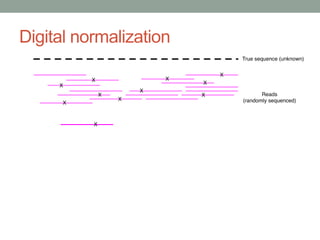
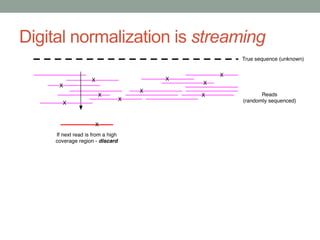
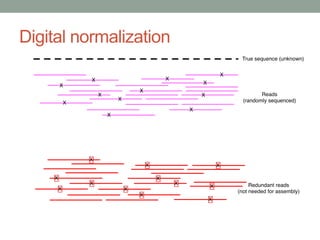
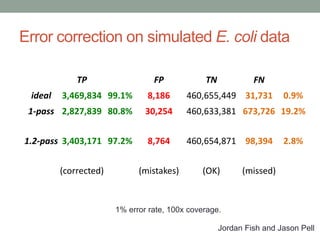
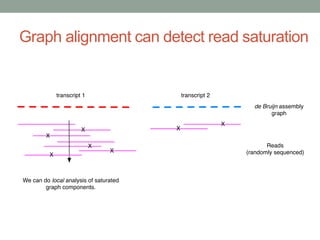
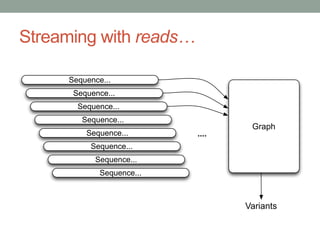
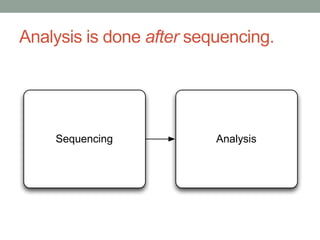
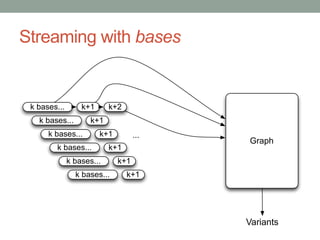
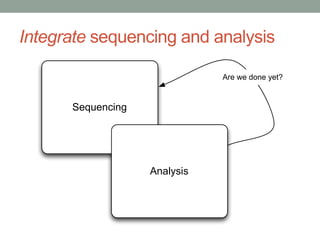
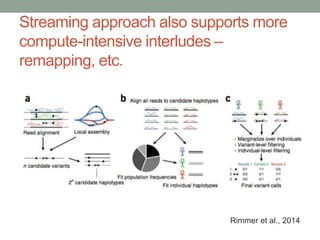
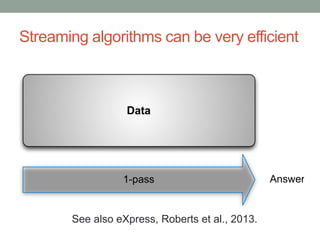
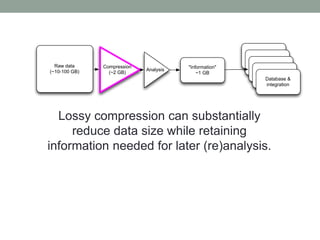
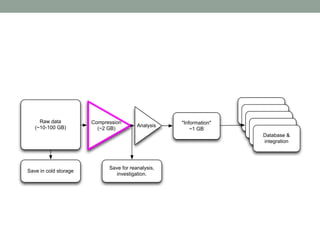
The document discusses potential approaches for streaming variant analysis of sequencing data as it is generated. It notes that current approaches are multi-pass and analyze data after sequencing. A streaming approach could integrate analysis with sequencing, processing data in a single pass as it is generated to call variants in real-time. This could avoid biases from referencing and enable early stopping if desired signals are detected. The approach relies on graph-based data structures and algorithms like digital normalization that compress data while retaining informative content.