Downloaded 478 times


































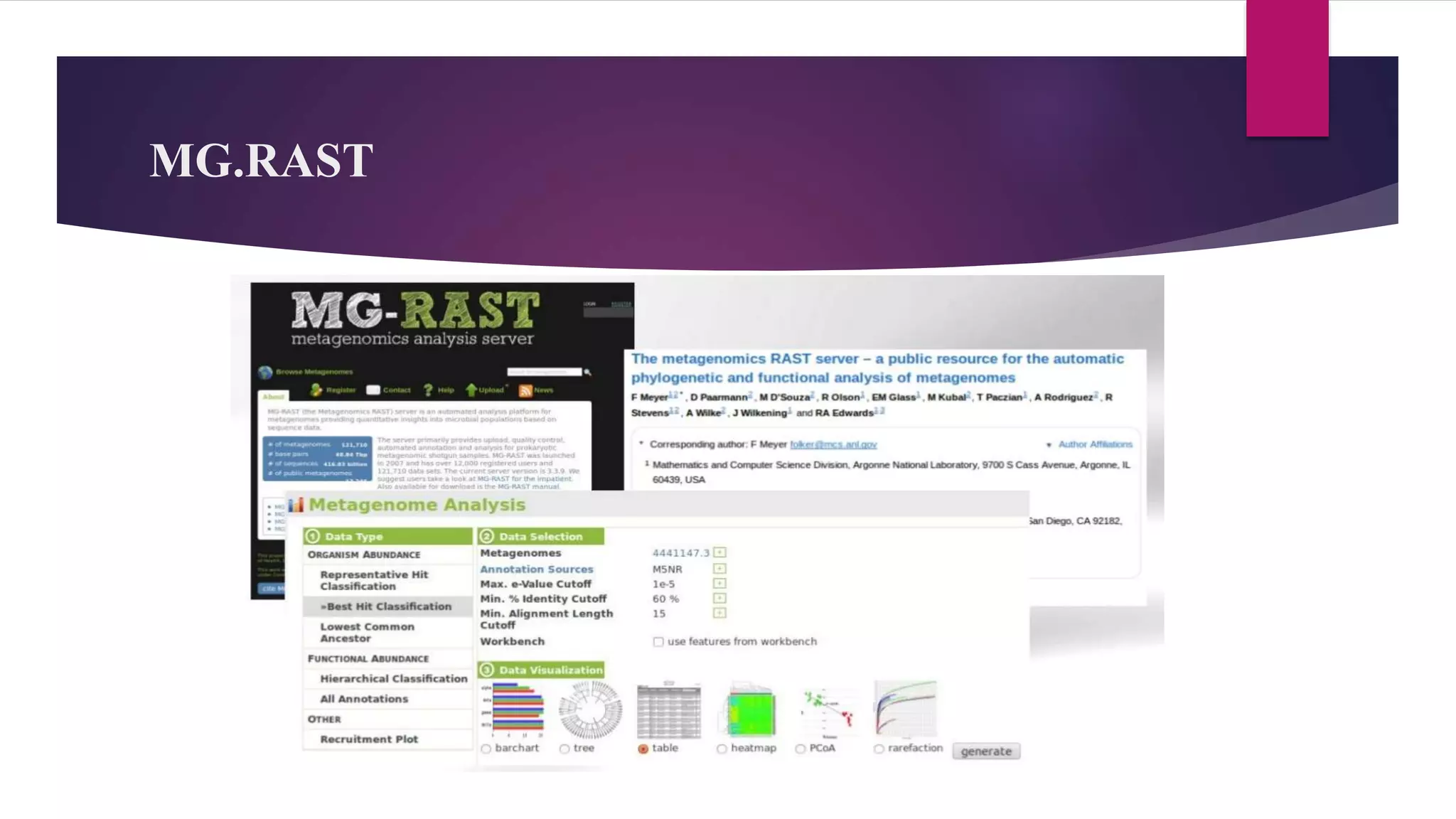
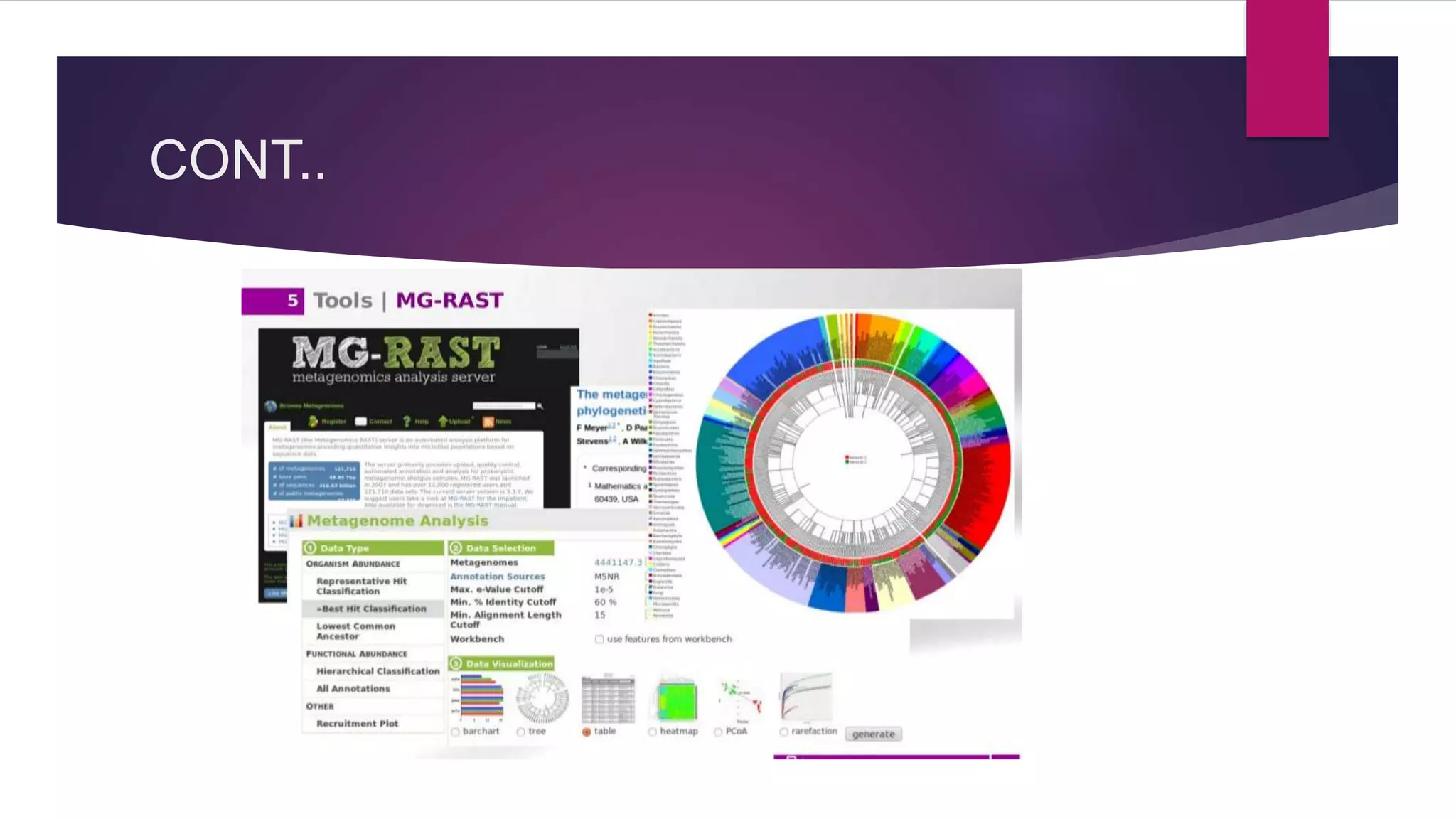




Shotgun metagenomics sequencing allows researchers to comprehensively sample all genes in organisms present in a complex sample without culturing. This provides insights into bacterial diversity, abundance, and uncultured microbes. Bioinformatics pipelines guide analysis including quality filtering, assembly, binning, gene finding, fingerprinting, and phylogeny/diversity modeling to understand communities. Metagenomics has applications in antibiotic/drug discovery, bioremediation, agriculture, human microbiome mapping, and more. Tools like QIIME, Mothur, MEGAN, and MG-RAST facilitate large-scale metagenomic analysis.
Metagenomics studies genetic material from environmental samples, differing from traditional microbiology.
Two approaches: sequence-driven analyzing DNA sequences vs. function-driven screening for specific functions.
Both metagenomic approaches face challenges in gene expression and reliance on existing knowledge.
Metagenomic data analysis begins with pre-filtering low-quality sequences to enhance data accuracy.
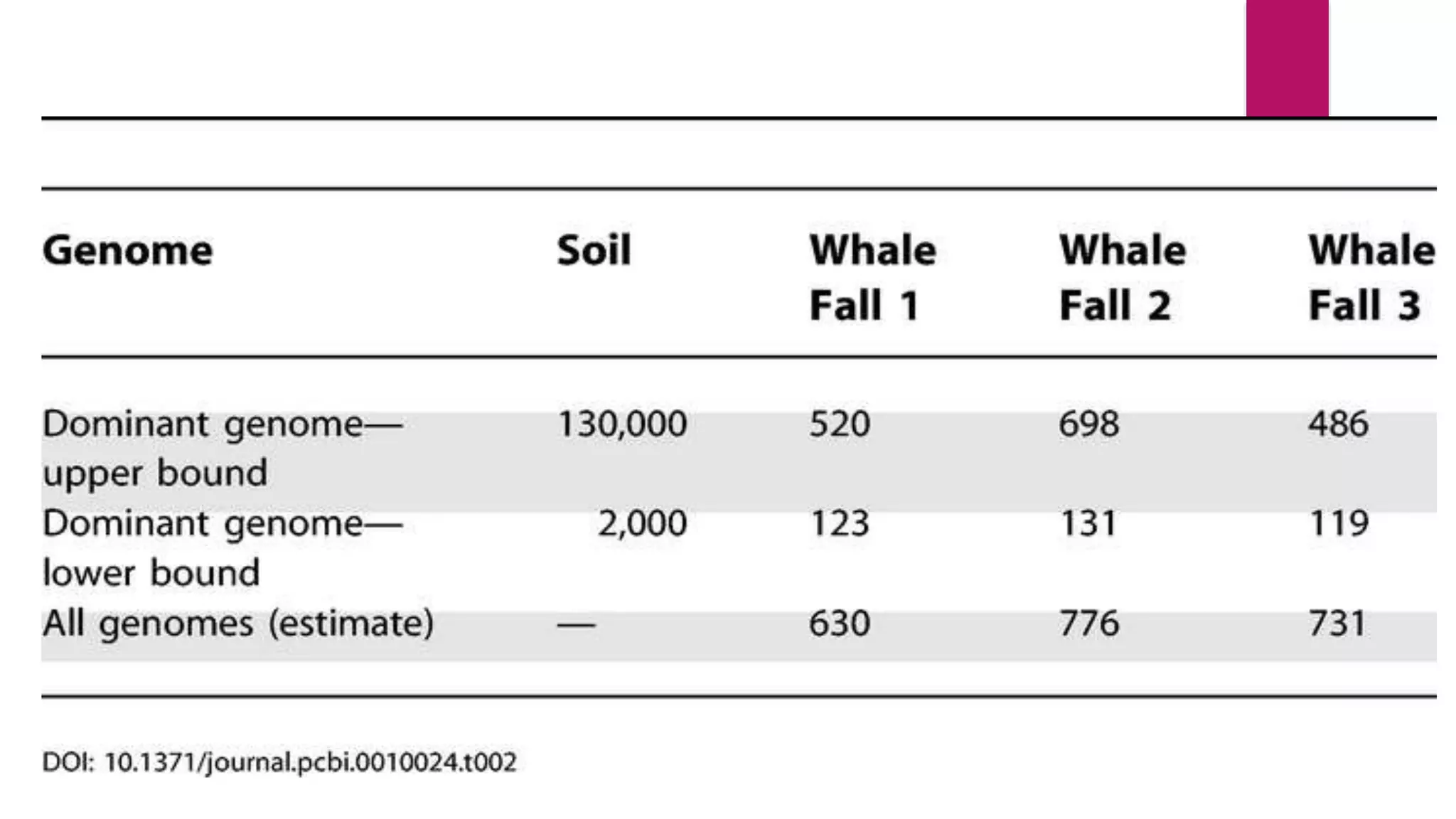
Comparison of metagenomes provides insights into microbial community functions influencing host health.
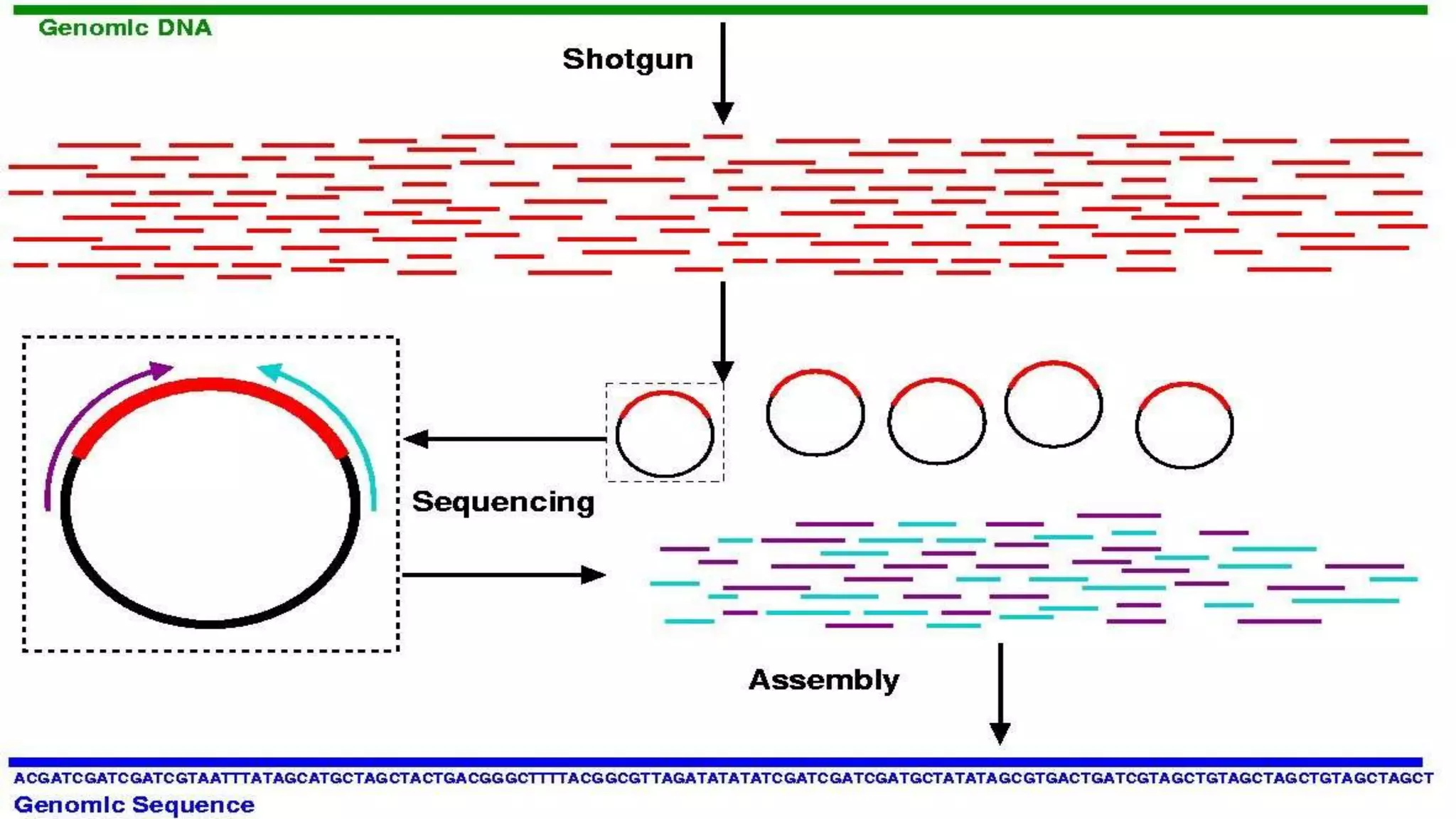
Shotgun sequencing breaks DNA into fragments for assembly, suitable for small genomes like viruses.
Faster and efficient than traditional methods, but requires a reference genome for accurate assembly.
High computational needs and assembly errors due to lack of genetic mapping are key disadvantages.
Assembly of microbial communities poses challenges due to unknown strain quantities and abundances.
WGS provides comprehensive gene sampling in microbiology, aiding in diversity evaluation of microbes.
Continued growth in shotgun sequencing projects promises to uncover ecological and community insights.
Technological advancements include A5-miseq and Orione for user-friendly bioinformatics data analysis.Metagenomics applications impact pharmaceuticals, environmental cleanup, and agriculture, showing diverse uses.
Focus on human microbiome mapping may yield tools for nutrition and understanding complex diseases.
QIIME, Mothur, and MEGAN are essential bioinformatics tools for analyzing microbial dynamics.
Future research will focus on exploring new antibiotics, studying gut microbiome effects and ancient DNA.






















































































