Downloaded 13 times






















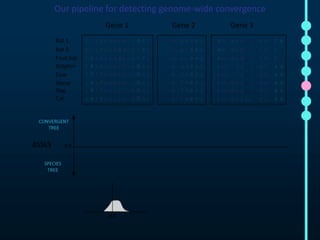
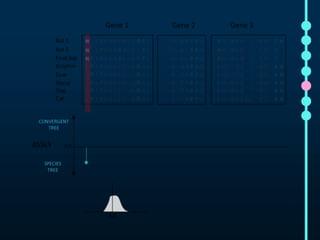
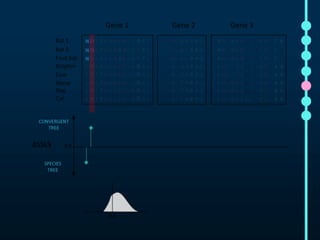
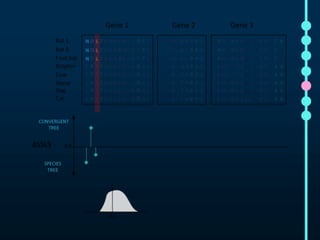
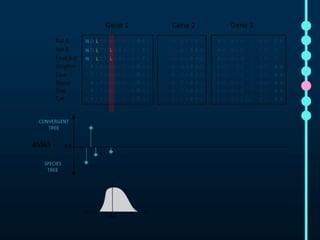
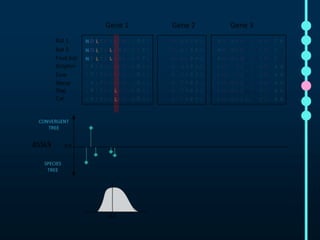
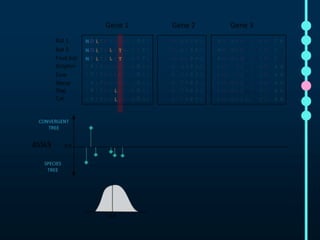
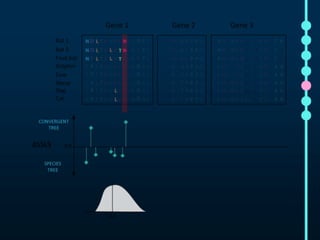
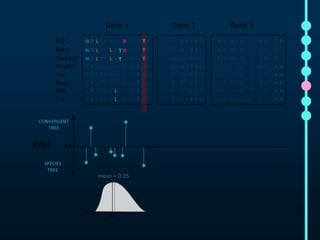
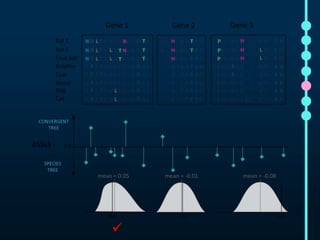










This document outlines Joe Parker's research interests in phylogenomics and high-throughput comparative genomics at Queen Mary University London. It discusses why phylogenomics is important, provides examples of past studies, and describes the lab's workflow and tools for sequencing, assembly, alignment, phylogeny inference, and phylogenetic analysis. It also presents a case study on detecting genome-wide convergence and discusses future directions including environmental metagenomics, cloud computing models, and real-time phylogenetics.








![[2013.10.29] albertsen genomics metagenomics](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131029070115-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)























































































