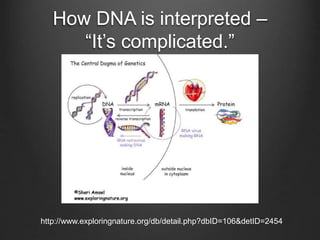
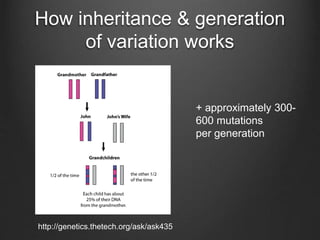
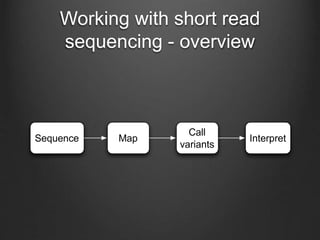
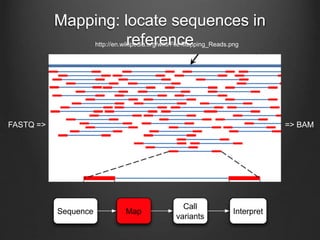
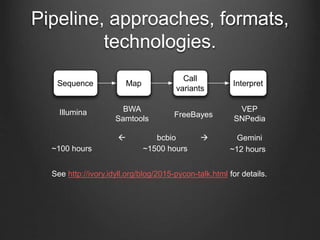
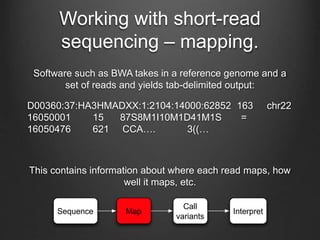
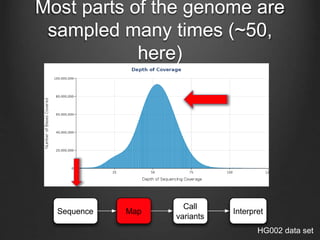
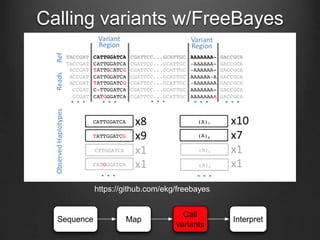
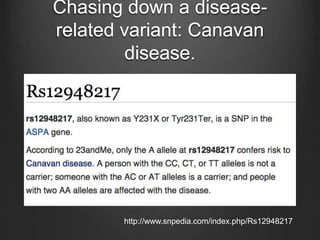
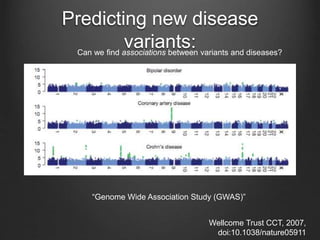
The document discusses how to interpret a person's genome sequence. It explains that while we can identify genetic variations and inherited conditions, a genome sequence alone does not reveal much because DNA is complex code that is difficult to decipher. Environmental factors and how genes interact also influence traits. The document outlines the process of genome sequencing, mapping reads to a reference, variant calling, and challenges in interpretation due to incomplete knowledge and versioning issues.