Downloaded 59 times



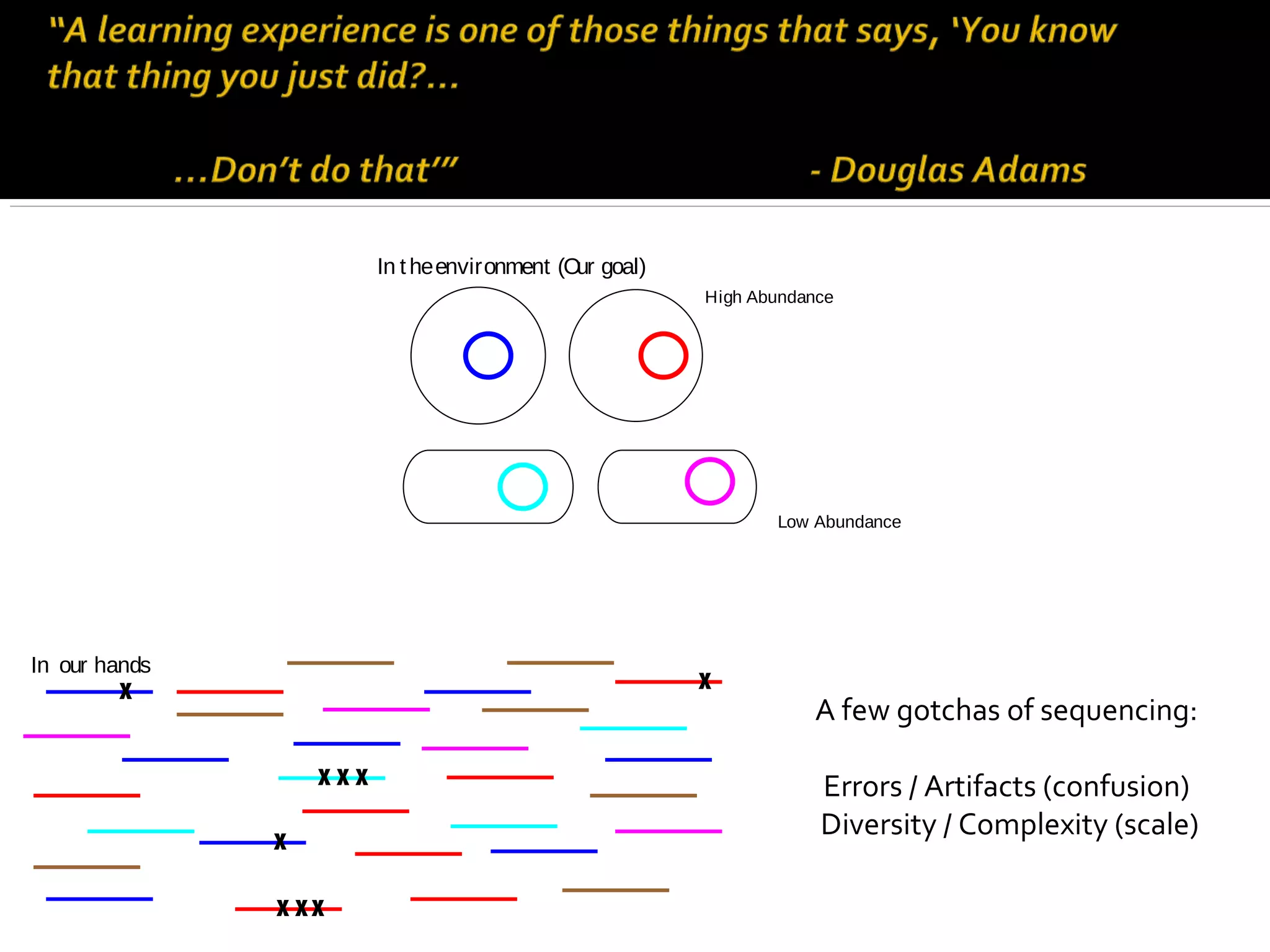
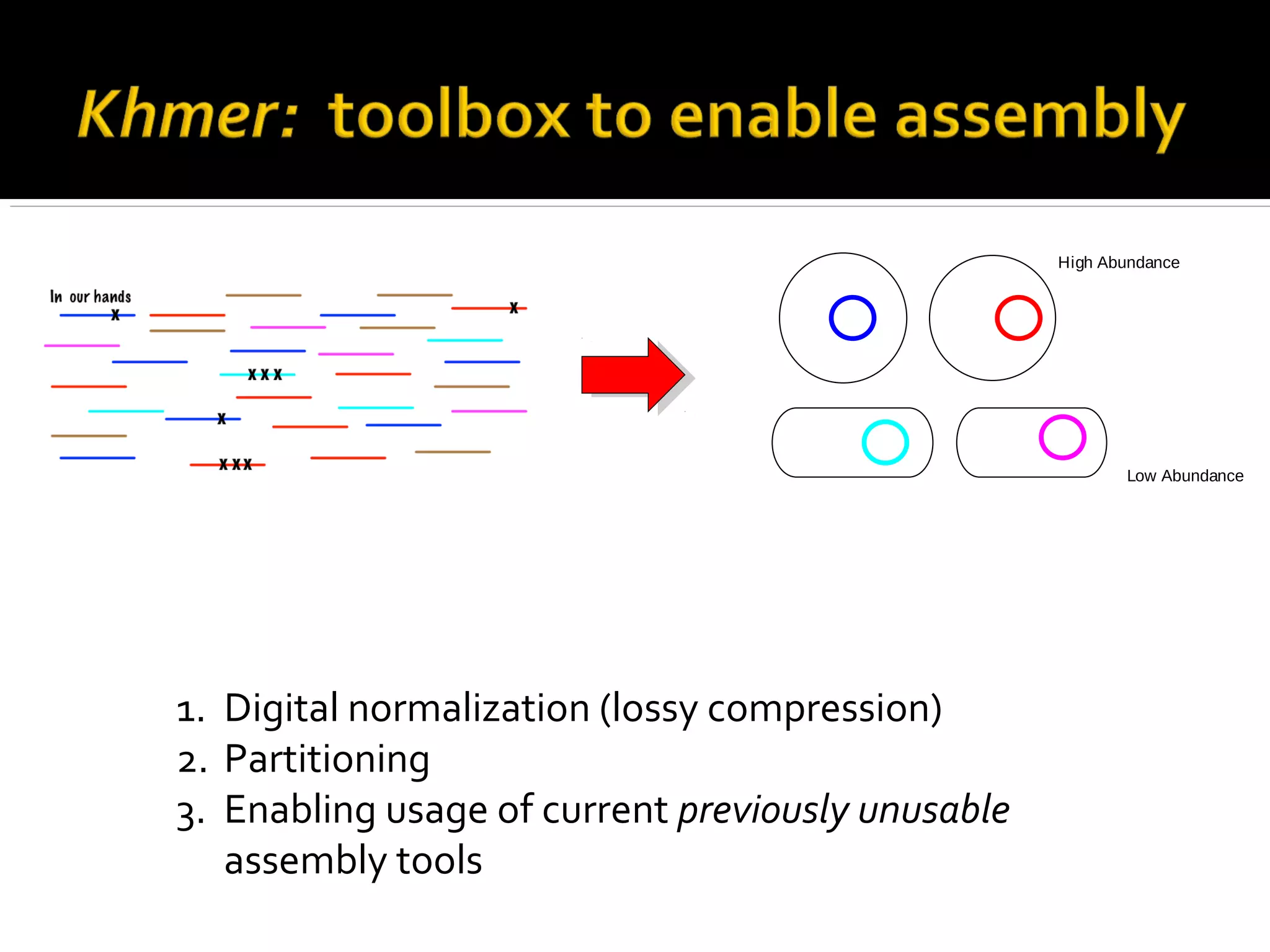

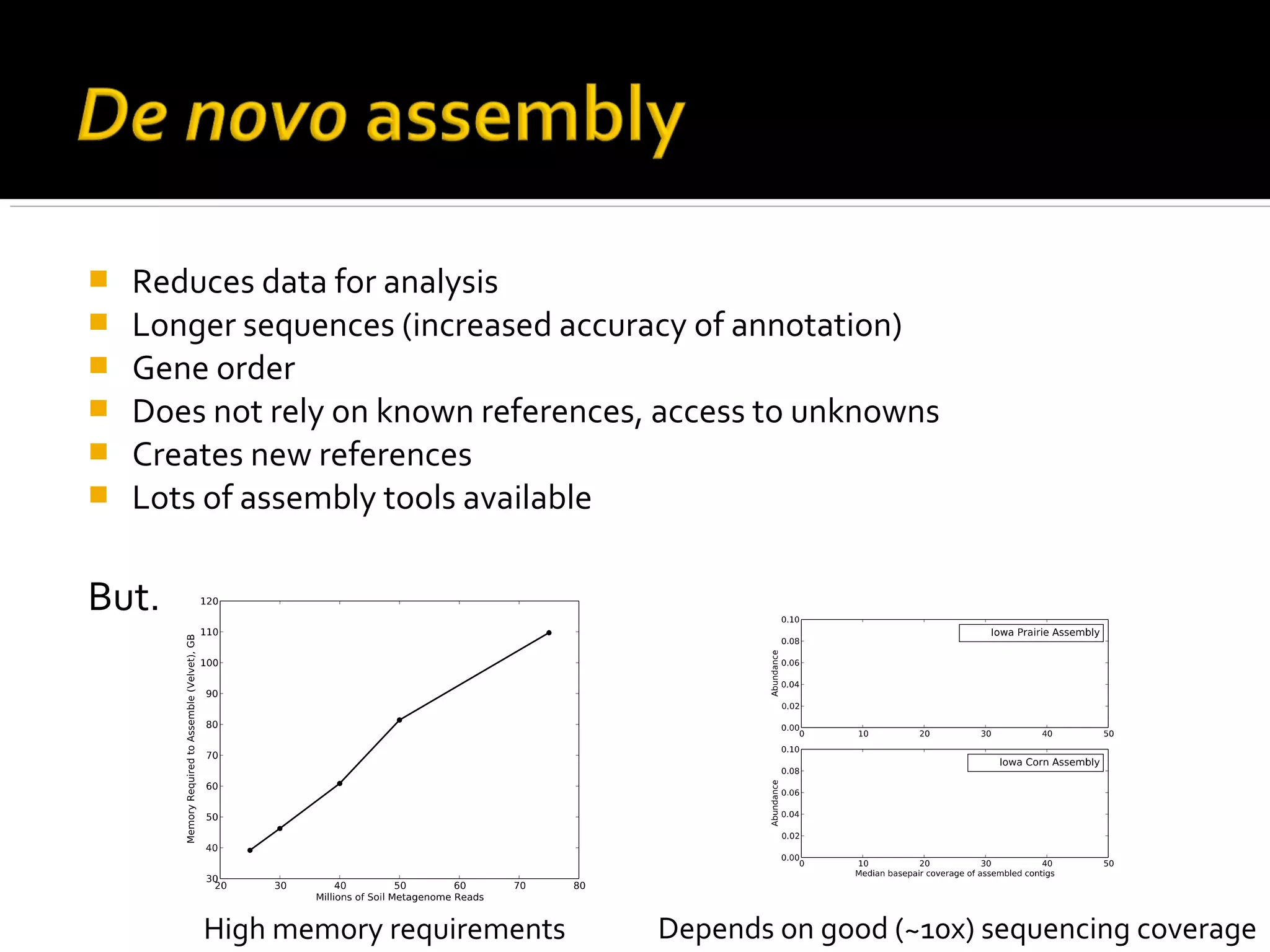

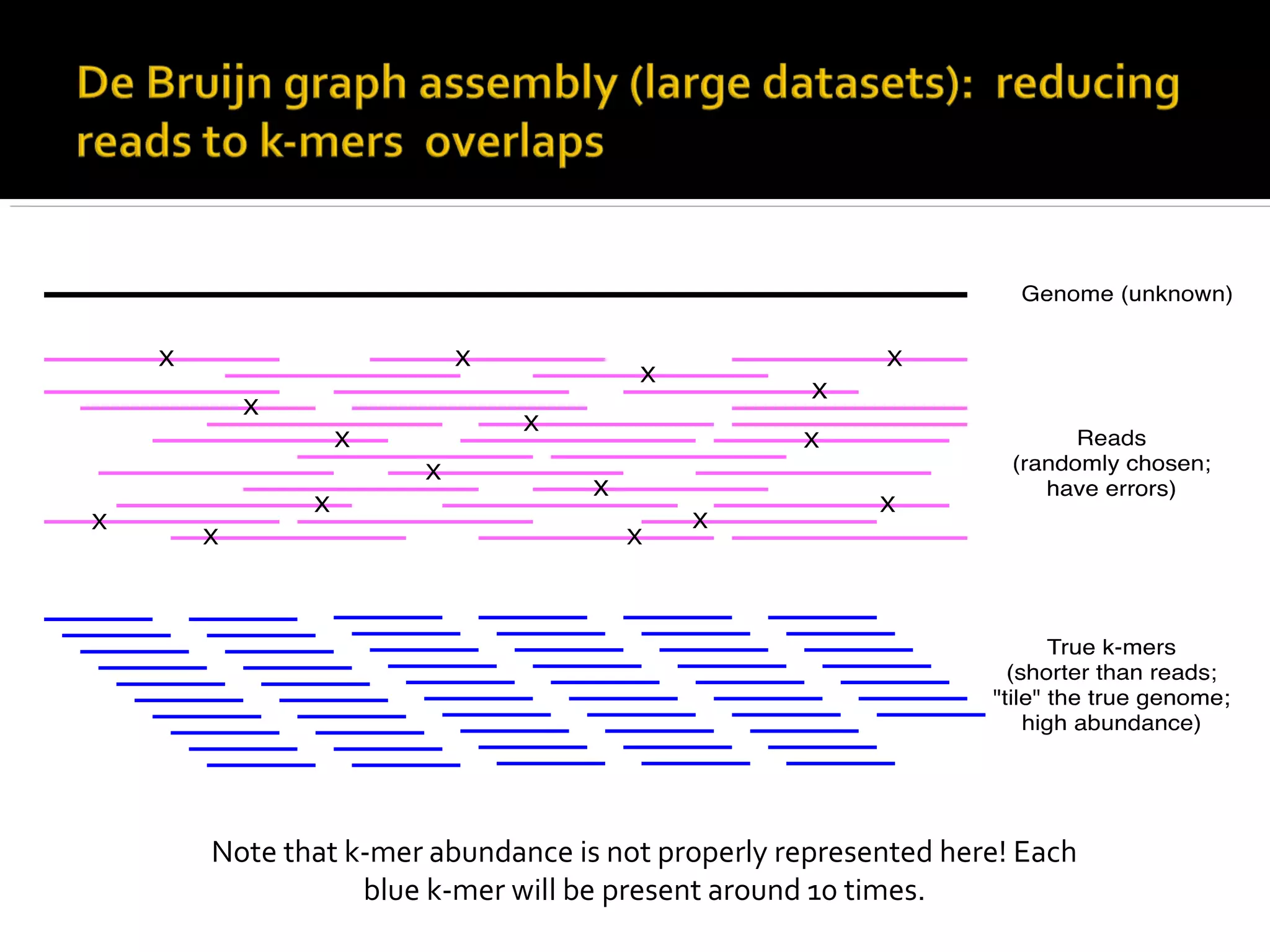
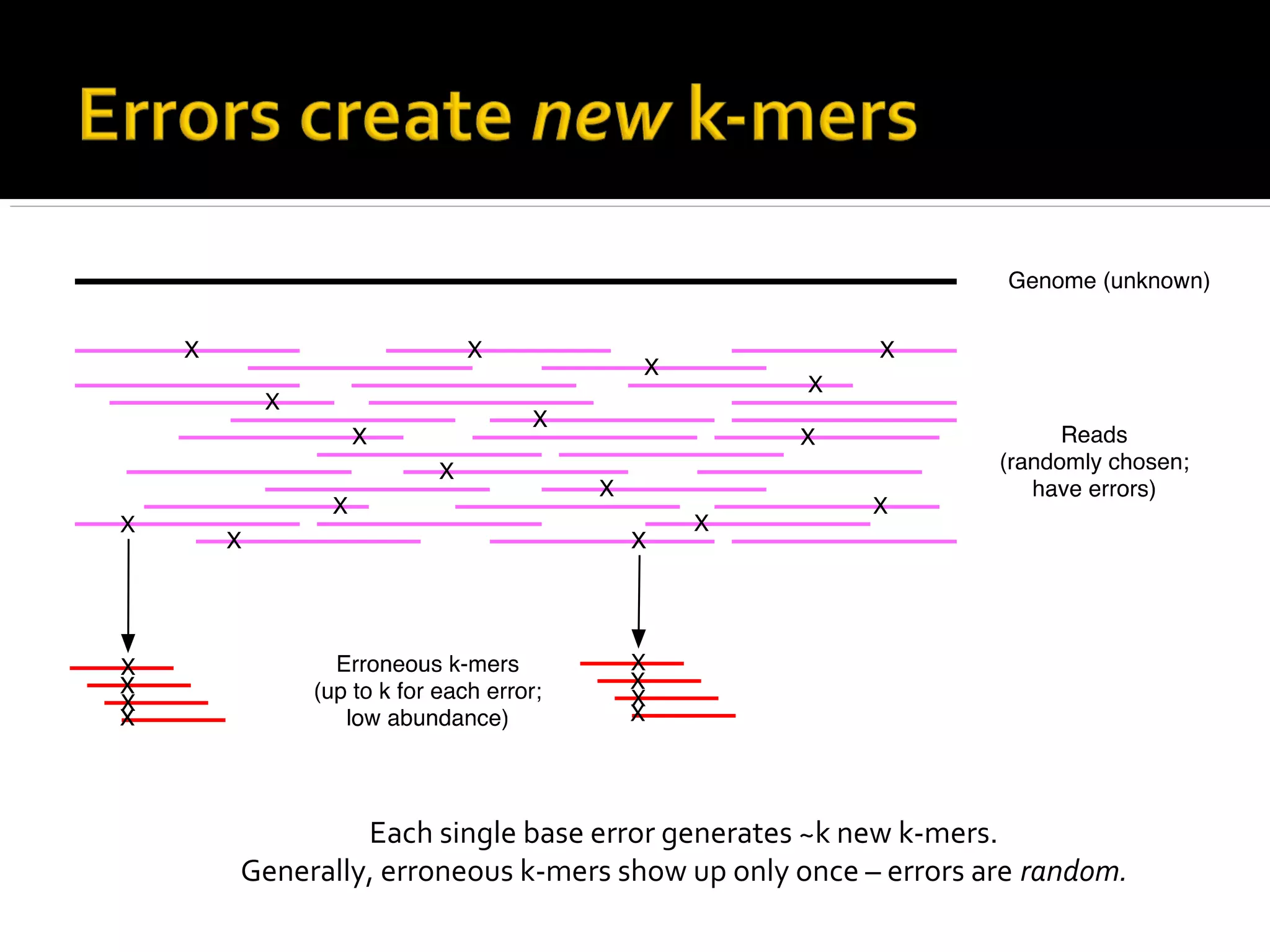
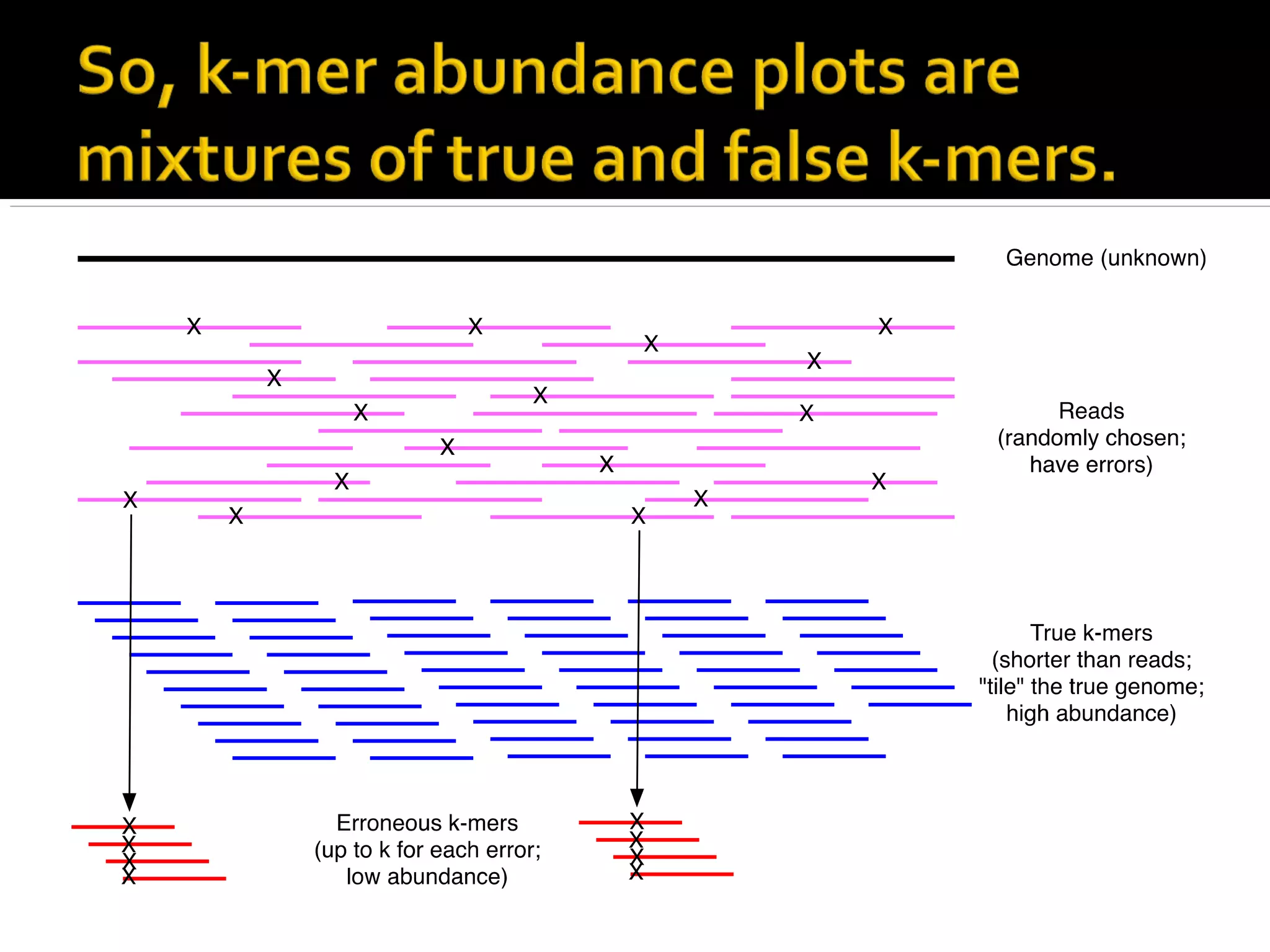

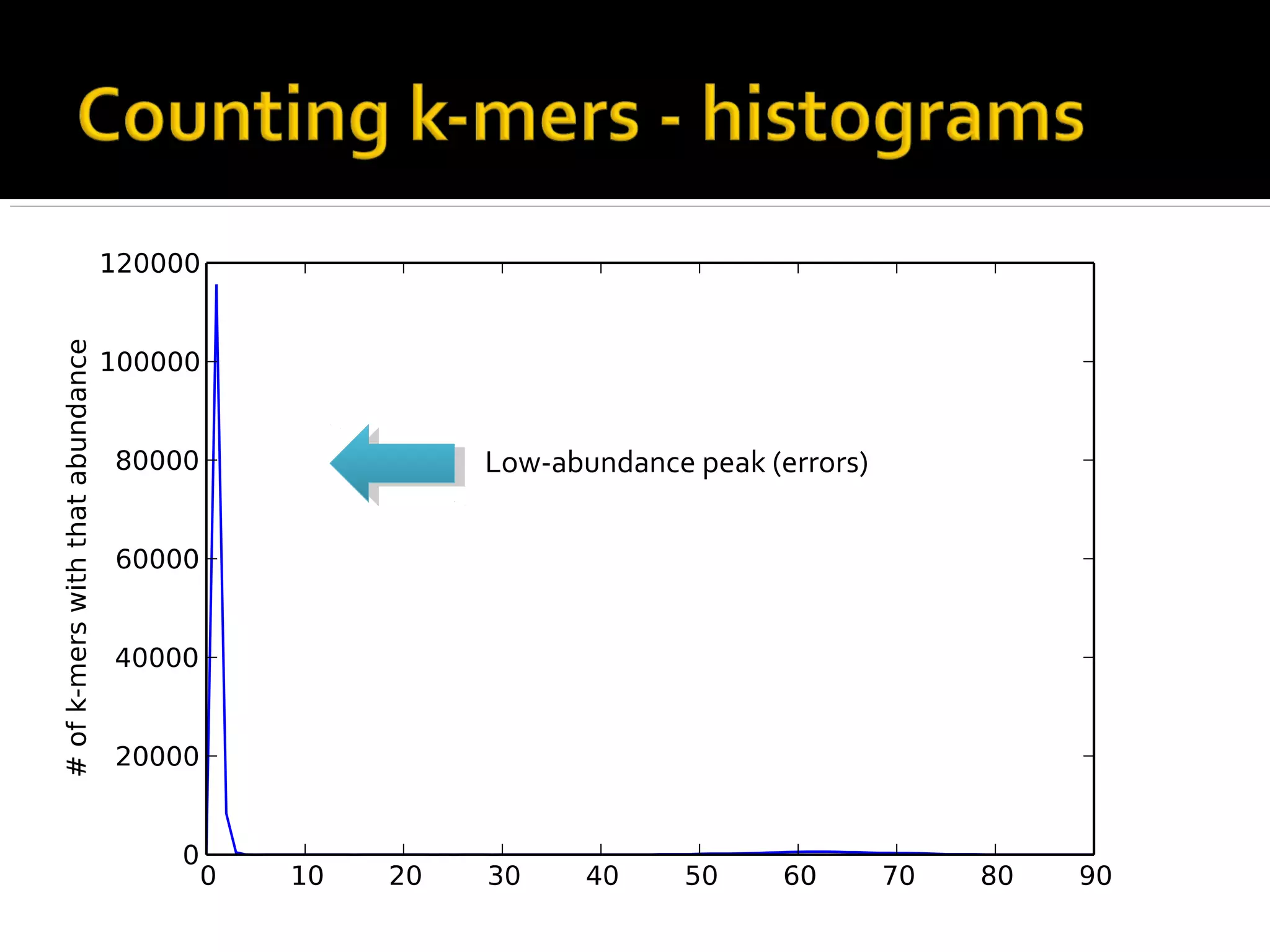
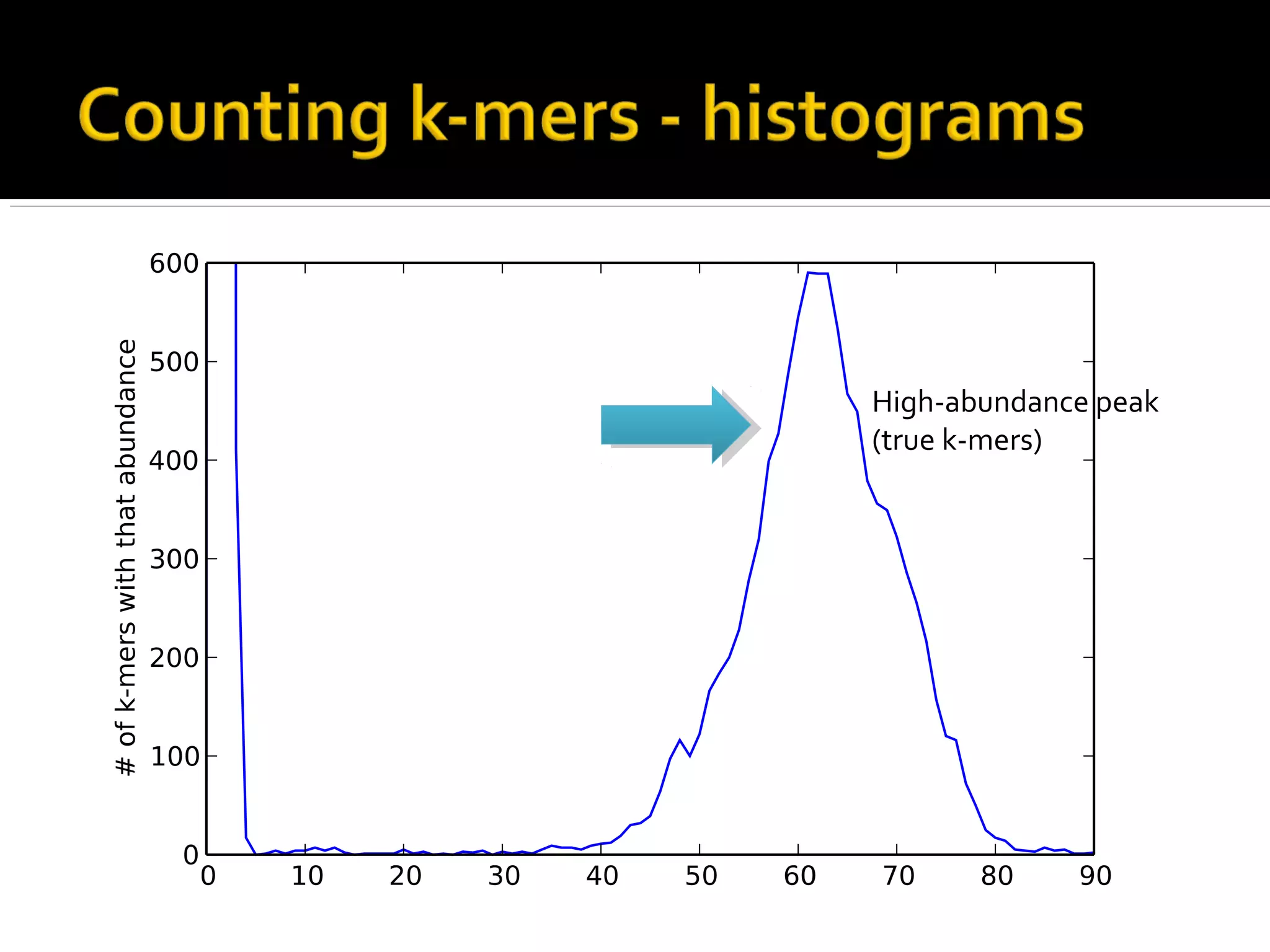



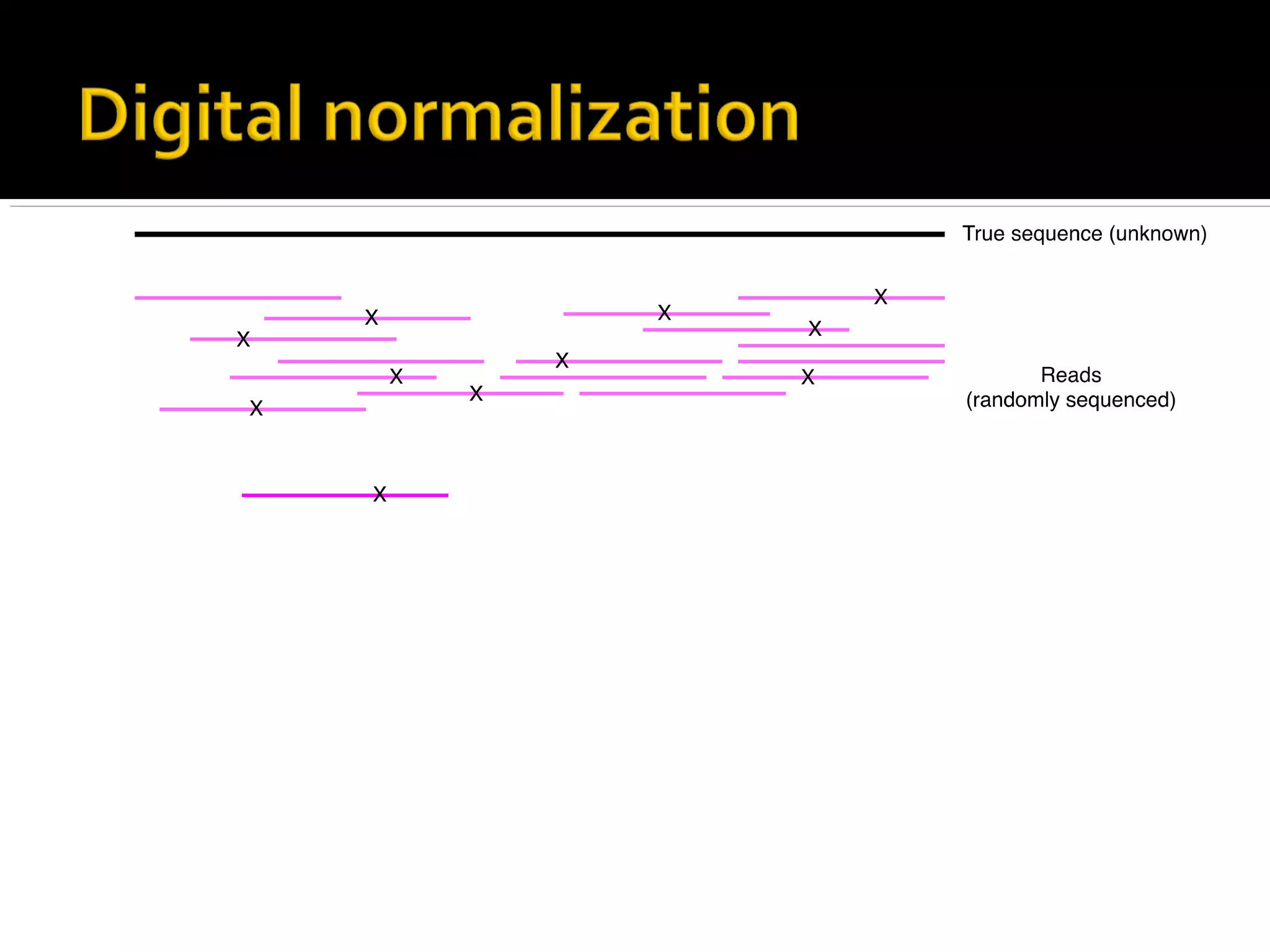

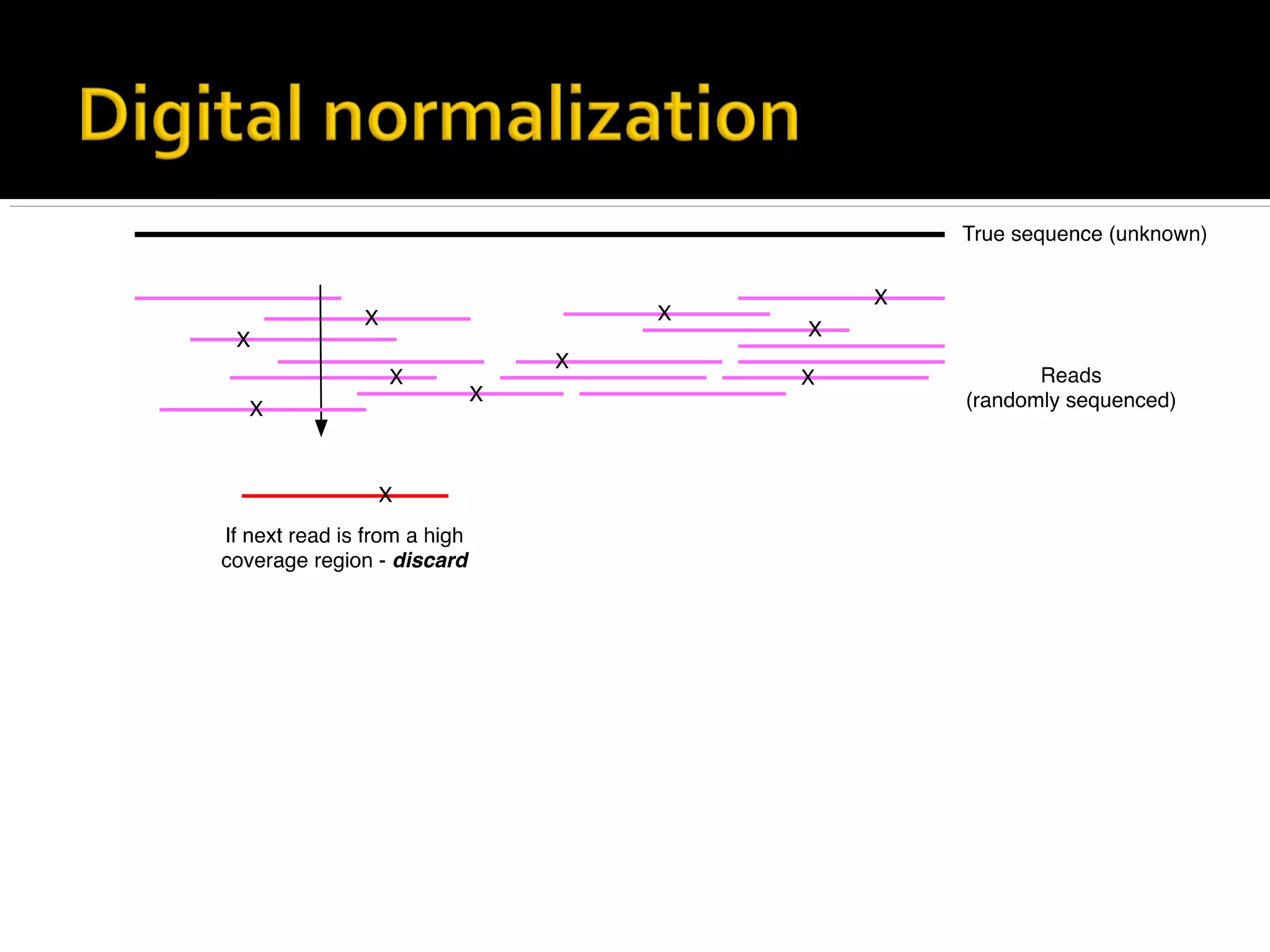
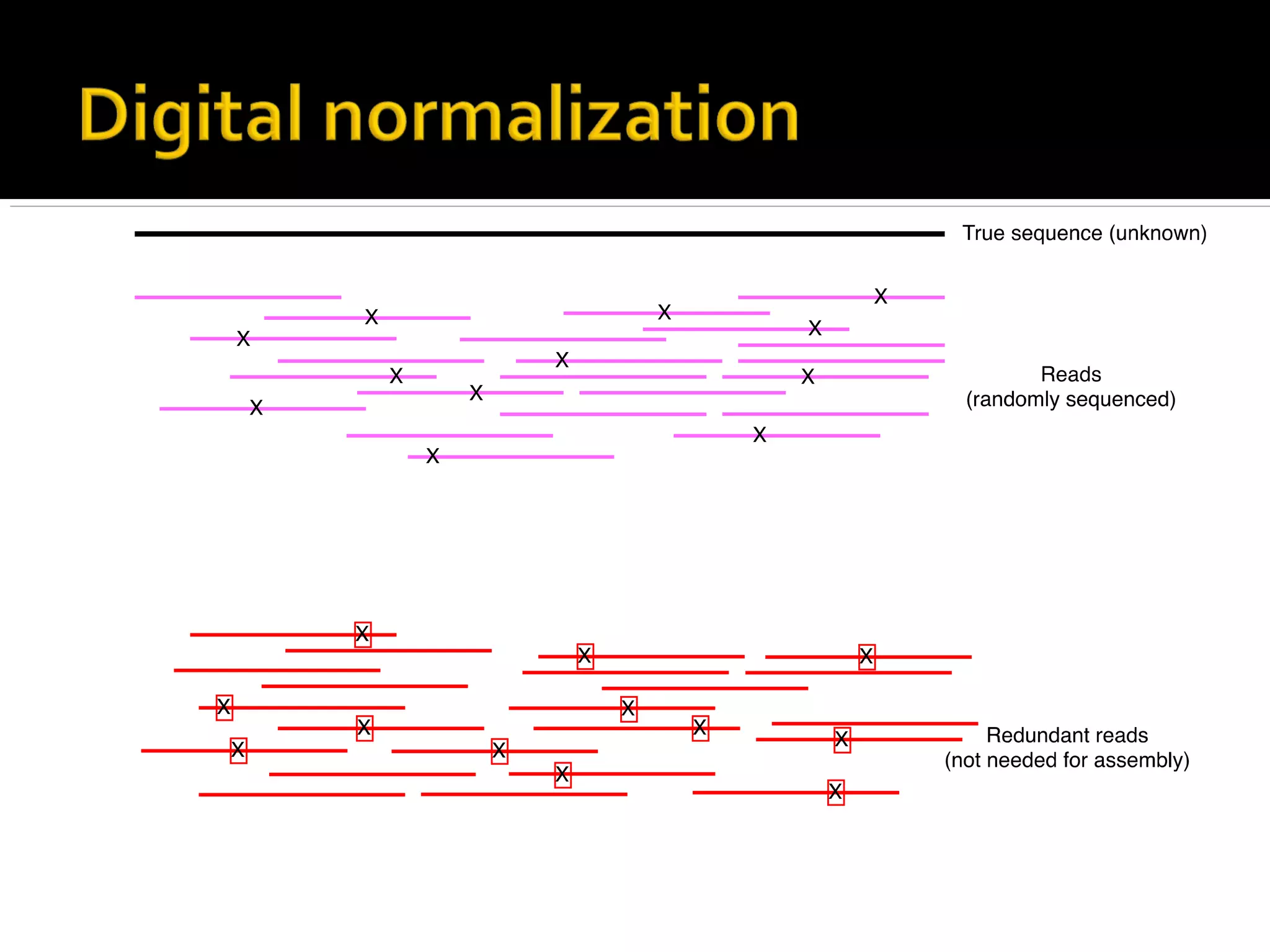


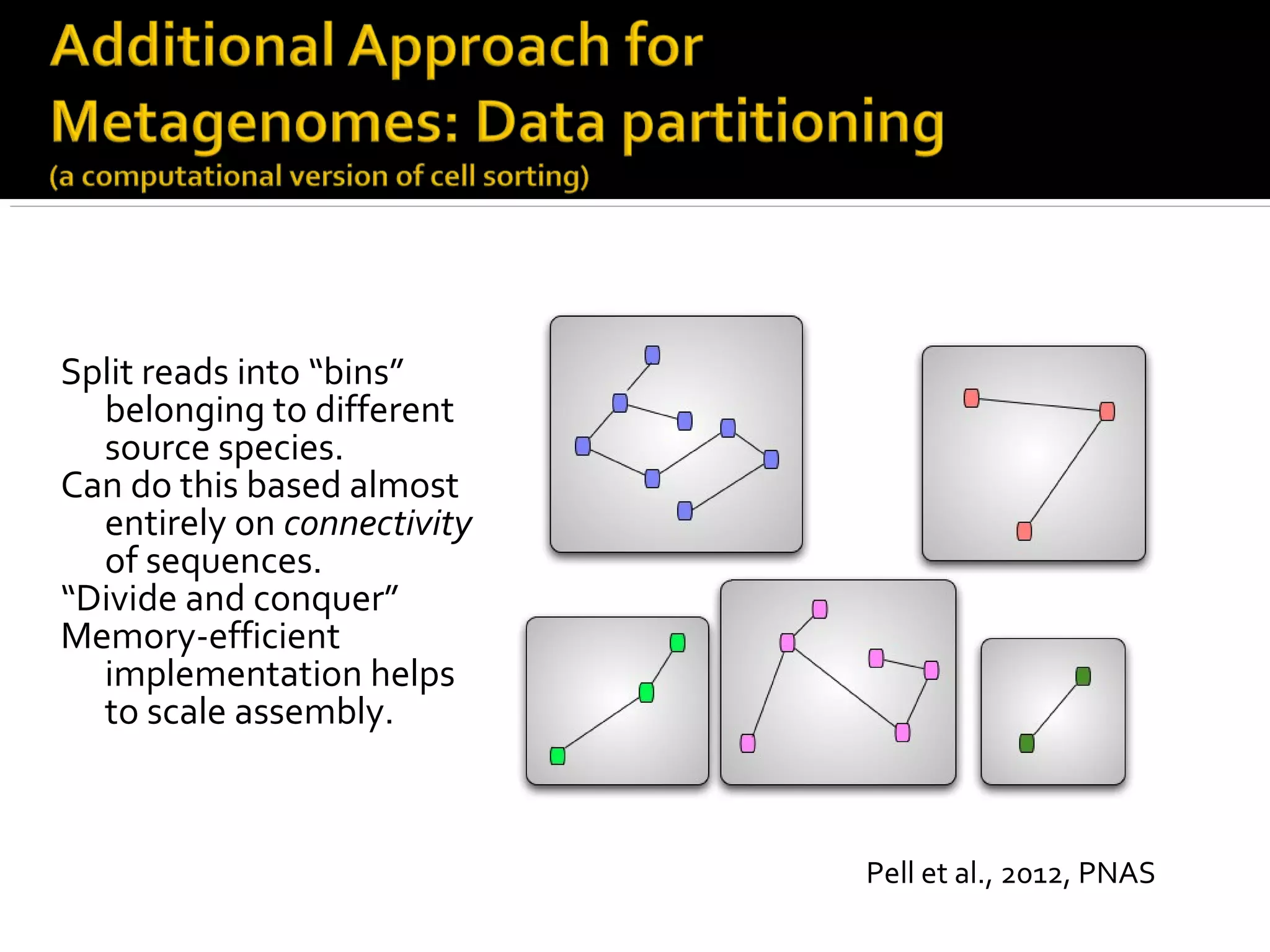

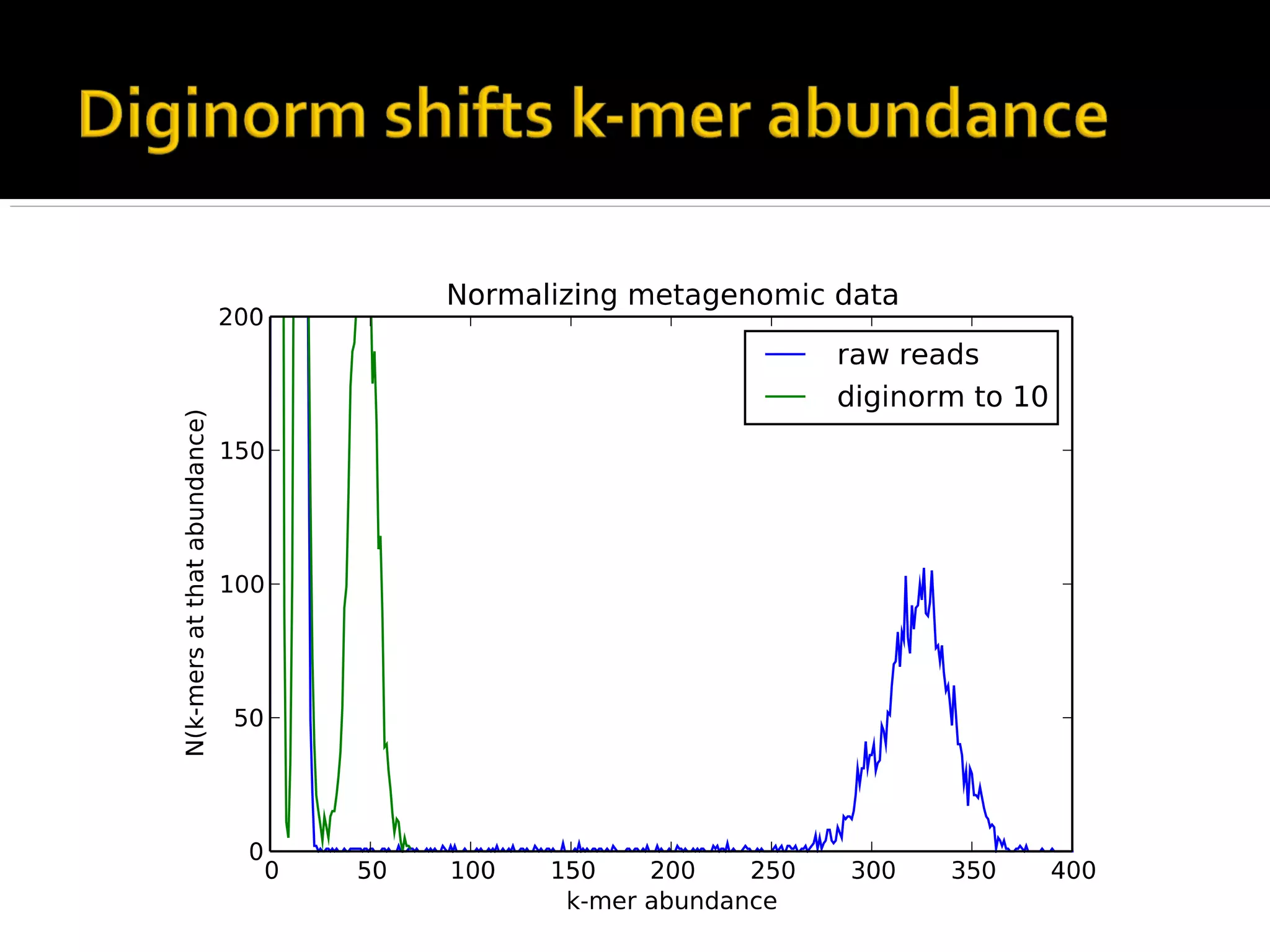
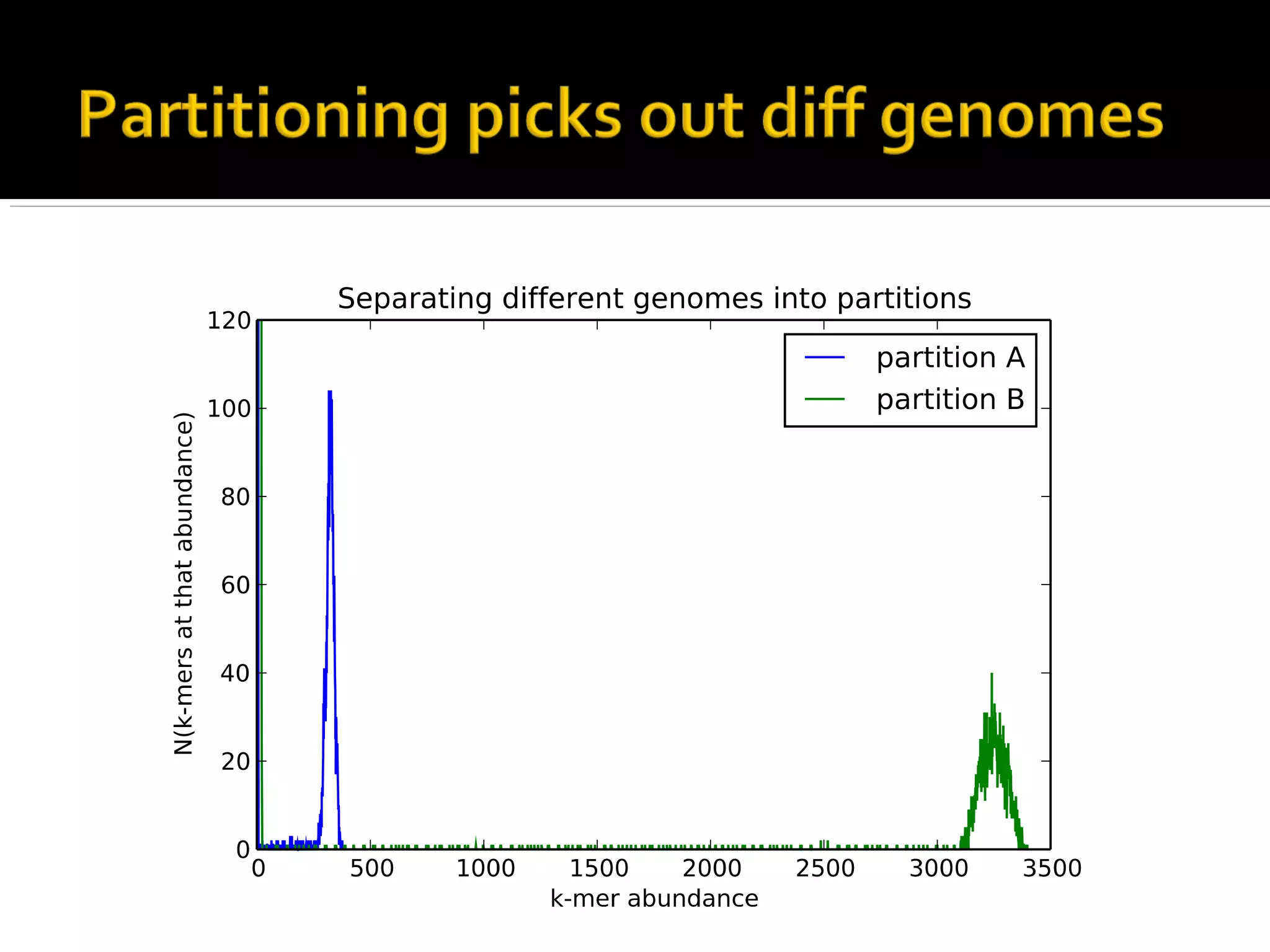














This document summarizes a presentation given by Adina Howe at the ASMWorkshop in May 2013 on visualizing complexity in metagenomics data. It lists her collaborators at Michigan State University and Argonne National Laboratory. It discusses challenges in sequencing metagenomics samples like errors, diversity, and low abundance of sequences. It describes techniques like digital normalization and partitioning that can help scale assembly of large metagenomics datasets. It addresses questions around memory requirements, evaluating assemblies, and studying microbial communities and environments.












































































