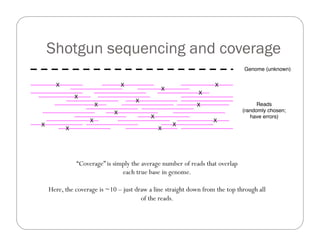
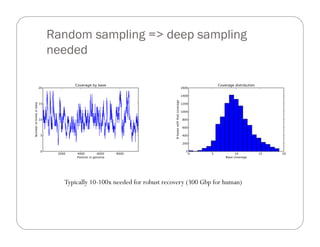
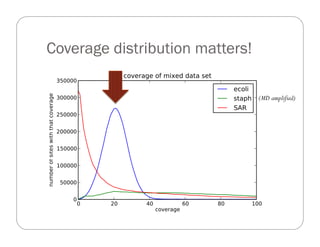
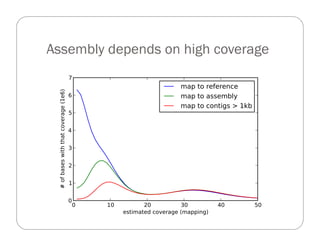
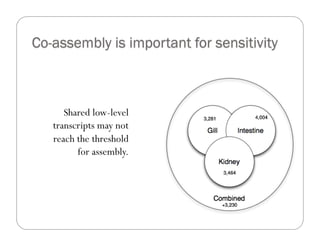
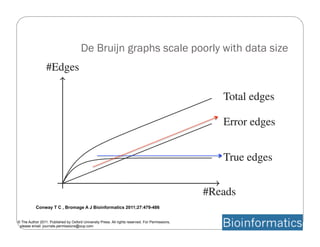
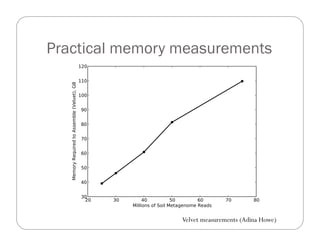
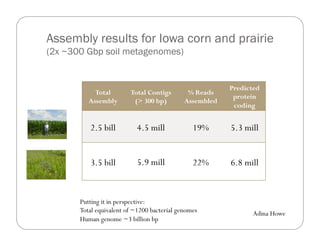
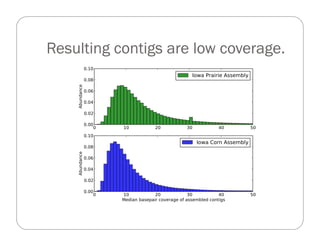
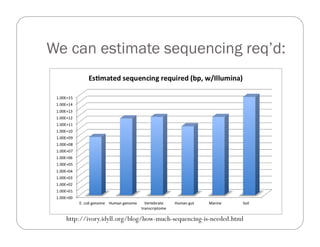
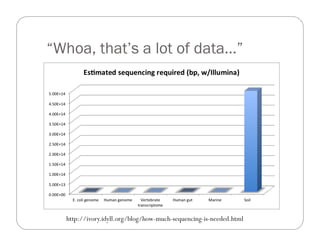
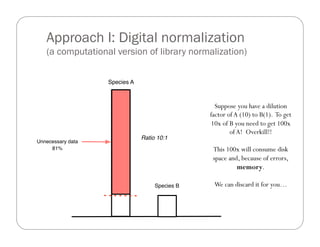
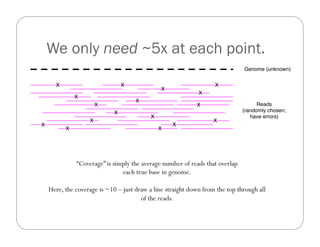
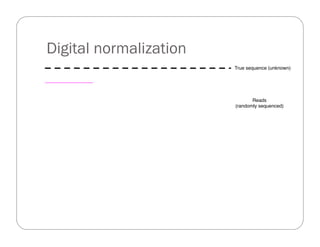
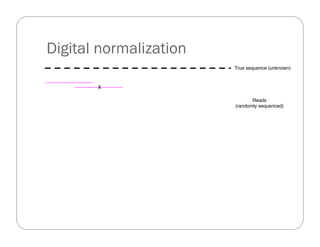
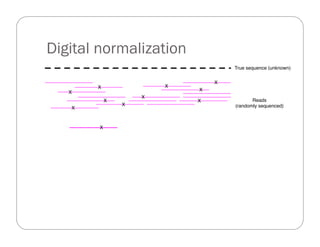
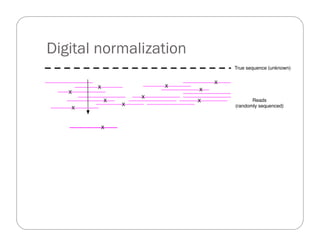
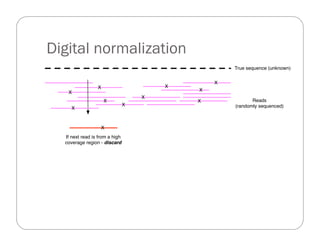
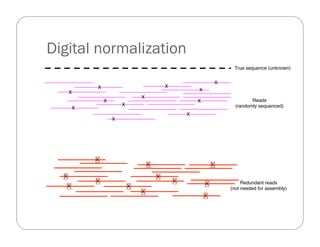
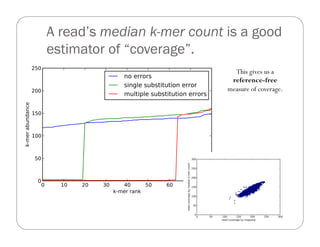
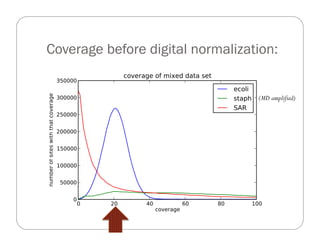
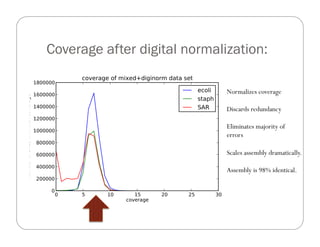
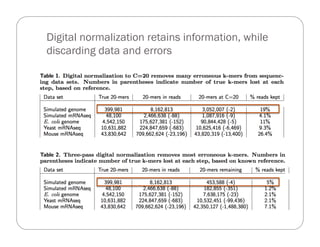
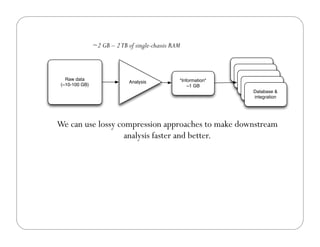
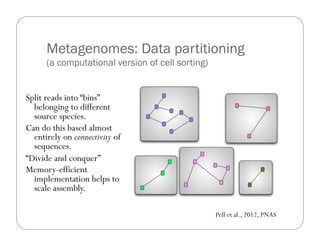
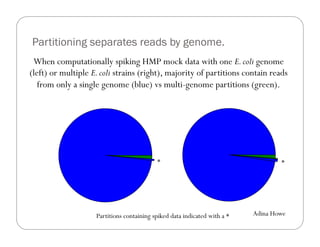
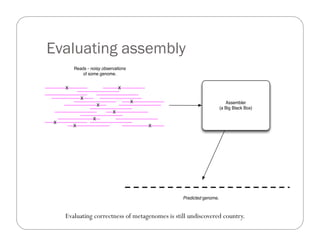
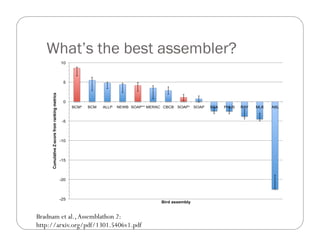
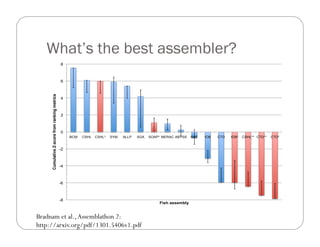
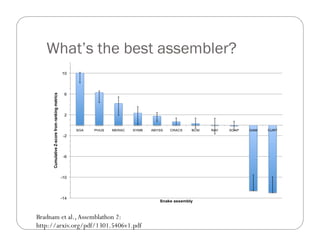
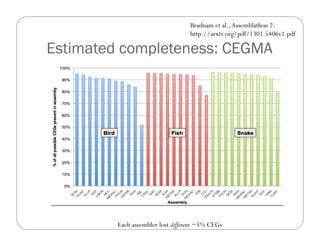
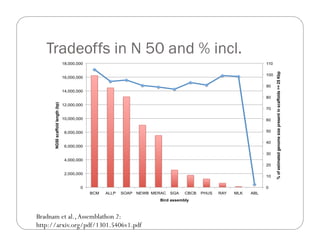
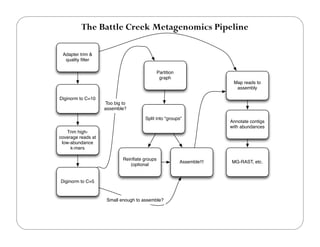
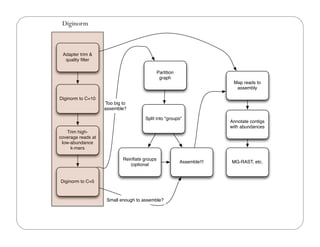
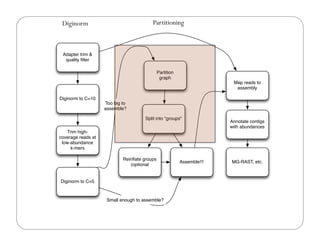
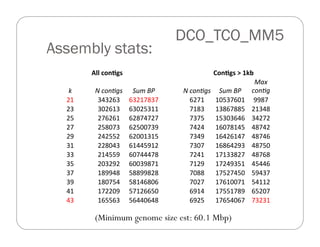
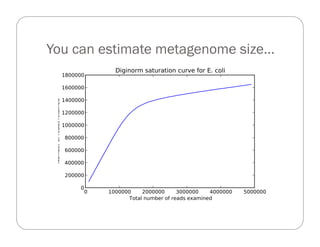
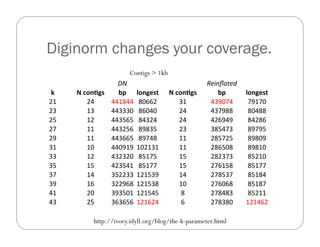
This document provides an overview of the Battle Creek Metagenomics Pipeline for assembling large, complex metagenomes. The pipeline first uses digital normalization techniques like Diginorm to equalize coverage and remove redundant reads. It then partitions the reads into "groups" to separate genomes. Smaller partitions are assembled, while larger ones are further processed. The result is contigs that can be annotated and analyzed. Key aspects of the pipeline are the use of digital normalization and partitioning methods to improve scaling and allow assembly of very large metagenomic datasets.