Downloaded 6,276 times





![Henderson-Hasselbach equation (clinically
relevant form)
• pH = pKa + log([HCO3-]/.03xpCO2)
• pH = 6.1 + log([HCO3-]/.03xpCO2)
• Shows that pH is a function of the RATIO
between bicarbonate and pCO2
• PCO₂ - ventilatory parameter (40 +/- 4)
• HCO₃⁻ - metabolic parameter (22-26 mmol/L)](https://image.slidesharecdn.com/acidbasebalancefinal-120325203702-phpapp02/85/Acid-base-balance-5-320.jpg)










![Acid Base Disorders
Disorder pH [H+] Primary Secondary
disturbance response
Metabolic [HCO3-] pCO2
acidosis
Metabolic [HCO3-] pCO2
alkalosis
Respiratory pCO2 [HCO3-]
acidosis
Respiratory pCO2 [HCO3-]
alkalosis](https://image.slidesharecdn.com/acidbasebalancefinal-120325203702-phpapp02/85/Acid-base-balance-26-320.jpg)



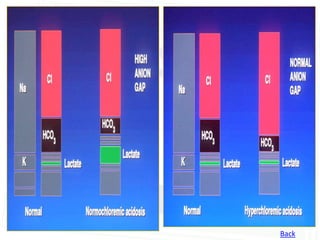





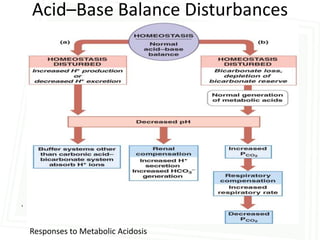





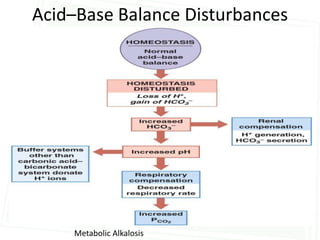






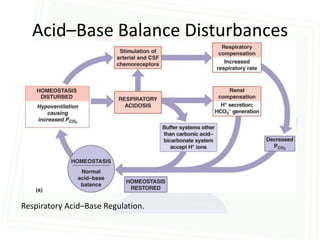
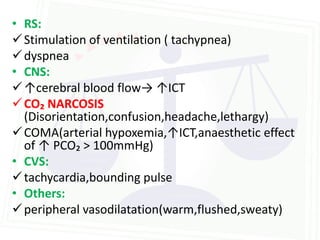




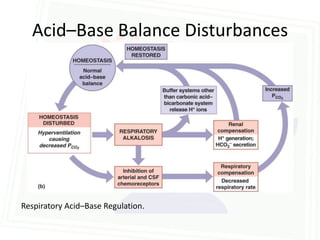
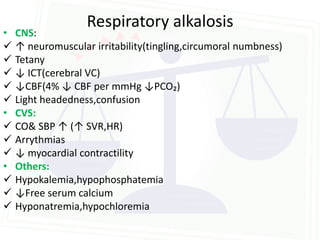
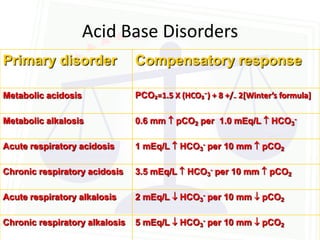



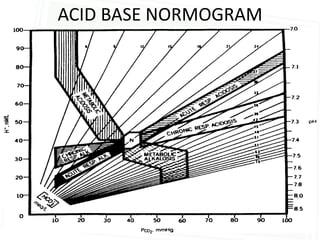



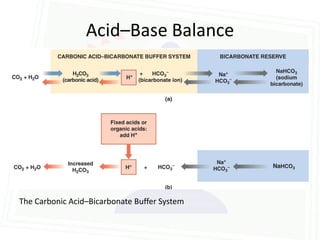
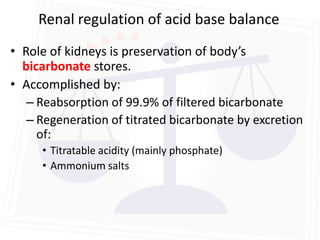
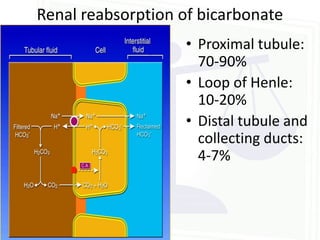
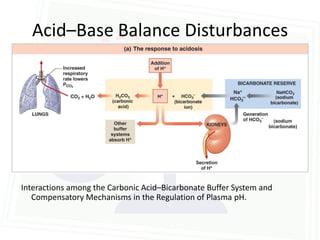
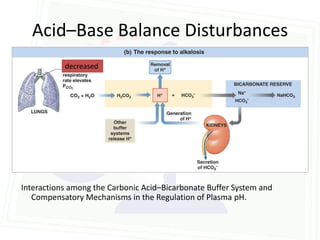
This document discusses acid-base balance and disorders. It begins by defining acids and bases, and describing the normal physiology of acid-base balance. It then discusses the four main types of acid-base disorders: metabolic acidosis, metabolic alkalosis, respiratory acidosis, and respiratory alkalosis. For each disorder it describes the primary disturbance (pH or HCO3-) and the secondary compensatory response. The document goes on to provide details on the causes, mechanisms, and clinical assessments of different metabolic and respiratory acid-base disorders.







































































































