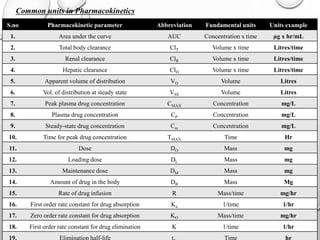
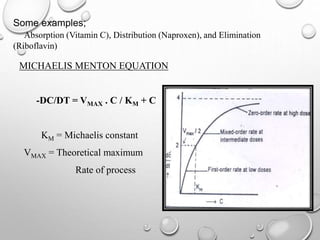
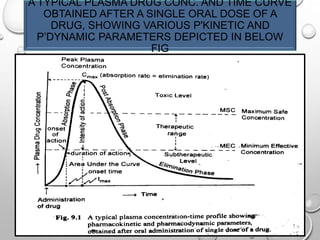
Downloaded 4,705 times




































![• At steady state. The rate of change of amount of drug in the body is zero ,eq
23 becomes
Zero=ro-kexss …27
Kexss=ro …28
Css=ro/kevd …29
=Ro/clt i.E infusion rate ....30
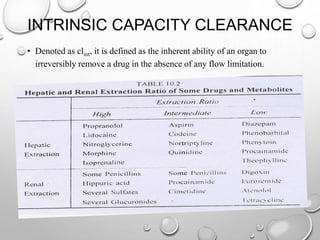
Clearance
Substituting eq. 30 in eq. 26
• C=css(1-e-ket) …31
Rearrangement yields:
• [Css-c]=e-ket
. ...32
Css
Log CSS-C = -ket …33
Css 2.303](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-45-320.jpg)
![• If n is the no. Of half lives passed since the start of infusion(t/t1/2)
• Eq. Can be written as
• C=CSS [1-(1/2)n] …34](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-46-320.jpg)


![ONE COMPARTMENT MODEL: EXTRA
VASCULAR ADMIN ( FIRST ORDER
ABSORPTION)
• Drug that enters the body by first order absorption process gets distributed in
the body according to one compartment kinetic and is eliminated by first
order process.
• The model can be depicted as follows and final equation is as follows
Blood & other
Body tissues
Drug at
site
Ka
KE
First order
absorption
elimination
C=Ka F Xo/Vd (Ka-KE) [e -Ket-e-Kat] …41](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-50-320.jpg)


![Extending the relationship X= vd C
Dcc = K21 xp – K12 xc – KE xc
Dt vp vc vc
X= Amt. Of drug in the body at any time t remaining to be eliminated
C=drug conc in plasma
Vd =proportionality const app. Volume of distribution
Xc and xp=amt of drug in C1 and C2
Vc and vp=apparent volumes of C1 and C2
= K12 xc – K21 xp
Vc vp On integration equation gives conc of drug in central and
peripheral compartments at any given time t
Cp = xo [( K21 – a)e-at + (K12 – b)e-bt]
Vc b – a a – b
Xo = iv bolus dose](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-54-320.jpg)
![• The relation between hybrid and microconstants is given as :
a + b = K12 + K21 + KE
A b = K21 KE
Cc = a e-at + be-bt
Cc=distribution exponent + elimination
exponent
A and B are hybrid constants for two exponents and can be resolved by graph
by method of residuals.
A = X0 [K21 - A] = CO [K21 – A]
VC B – A B – A
B = X0 [K21 - B] = CO [K21 – B]
VC A – B A – B
CO = Plasma drug concentration immediately after i.v. Injection](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-55-320.jpg)



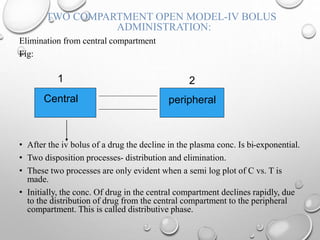
![TWO – COMPARTMENT OPEN MODEL- I.V.
INFUSION
The plasma or central compartment conc of a drug when administered as constant rate (0 order) i.V. Infusion is
given as:
C = R0 [1+(KE - b)e-at +(KE - a)e-bt]
VCKE b – a a - b
At steady state (i.E.At time infinity) the second and the third term in the bracket becomes zero and the equation
reduces to:
Css = R0
Vc ke
Now VC KE = vd b
Css = r0 = r0
Vdb clt
The loading dose X0,L = css vc = R0
Ke
1
Central
2
Peripheral](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-59-320.jpg)




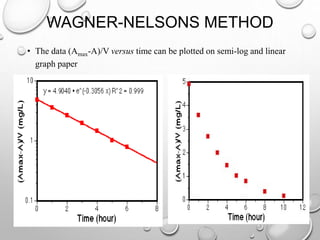
![WAGNER-NELSONS METHOD
• Example data for the method of wagner-nelson kel (from IV data) = 0.2 hr-
Time
(hr)
Plasma
Concentratio
n
(mg/L)
Column
3
ΔAUC
Column
4
AUC
Column 5
kel * AUC
A/V
[Col2 +
Col5]
(Amax - A)/V
0.0 0.0 0.0 0.0 0.0 0.0 4.9
1.0 1.2 0.6 0.6 0.12 1.32 3.58
2.0 1.8 1.5 2.1 0.42 2.22 2.68
3.0 2.1 1.95 4.05 0.81 2.91 1.99
4.0 2.2 2.15 6.2 1.24 3.44 1.46
5.0 2.2 2.2 8.4 1.68 3.88 1.02
6.0 2.0 2.1 10.5 2.1 4.1 0.8
8.0 1.7 3.7 14.2 2.84 4.54 0.36
10.0 1.3 3.0 17.2 3.44 4.74 0.16
12.0 1.0 2.3 19.5 3.9 4.9 -
∞ 0.0 5.0 24.5 4.9 4.9 -](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-64-320.jpg)


![φ It is commonly used in pharmacokinetics to resolve a
multiexponential curve into its individual components.
φ For a drug that follows one-compartment kinetics and
administered extravascularly, the concentration of drug
in plasma is expressed by a biexponential equation.
C=
퐾푎퐹푋0
[e-K
푉푑(퐾푎−퐾퐸)
t – e-K
E
a
t] (1)
If KaFX0/Vd(Ka-KE) = A, a hybrid constant, then:
C = A e-KEt – A e-Kat (2)](https://image.slidesharecdn.com/pharmacokineticmodels-140930004231-phpapp01/85/Pharmacokinetic-models-68-320.jpg)


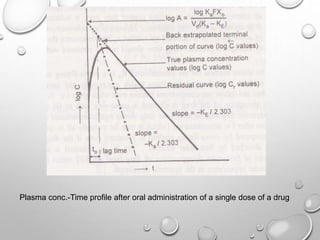




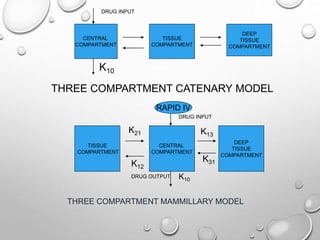




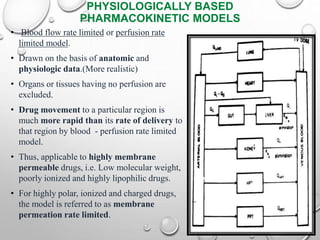





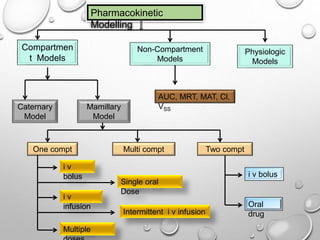
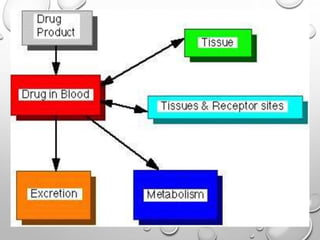
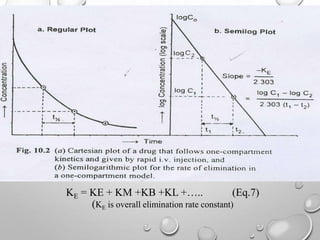
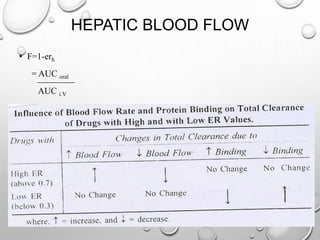
This document discusses pharmacokinetic models used to mathematically represent how drugs move through the body over time. It covers one compartment models, which assume rapid equilibrium between blood and tissues. For intravenous bolus administration, drug concentration decreases exponentially according to first-order kinetics. Key parameters include elimination rate constant, half-life, volume of distribution, and clearance. Compartmental modelling is useful for predicting drug concentrations, determining dosing schedules, and understanding drug interactions.























































































