Downloaded 9,282 times





![Factors affecting Bioavailability of a Drug
Physical properties of a drug
Physical state:
• Liquids > Solids
[ Solution > Suspension > Capsule > Tablet > Coated tablet ]
• Crystalloids > Colloids
Lipid or water solubility:
• Aqueous phase at absorption site
• Passage across Cell surface](https://image.slidesharecdn.com/kunalseminar-bioavailabilityandbioequivalence-130618125726-phpapp01/85/Bioavailability-and-Bioequivalence-Studies-10-320.jpg)






































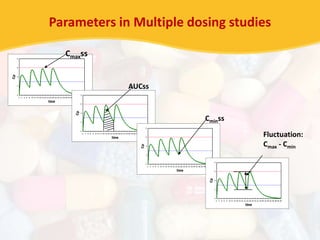

![Statistical Evaluation
Primary concern of bioequivalence is to limit Consumer’s &
Manufacturer’s risk
• Cmax & AUC analysed using ANOVA
• Tmax analysed by non-parametric methods
Use natural log transformation of Cmax and AUC
• Calculate Geometric means of Cmax of Test [Cmax’t]
• Calculate Geometric means of Cmax of Reference [Cmax’r]
• Calculate Geometric Mean Ratio= [Cmax’t] / [Cmax’r]
Calculate 90% confidence interval for this GMR for Cmax
Similarly calculate GMR for AUC](https://image.slidesharecdn.com/kunalseminar-bioavailabilityandbioequivalence-130618125726-phpapp01/85/Bioavailability-and-Bioequivalence-Studies-59-320.jpg)








![ Volunteer Selection & Recruitment
Volunteers called 1 day before study & admitted
Written ICF taken
During the Study
Standardized study environment
Vital signs examination at scheduled times
Standardised amount of water [~240ml]
No concomitant medications [including herbal remedies]](https://image.slidesharecdn.com/kunalseminar-bioavailabilityandbioequivalence-130618125726-phpapp01/85/Bioavailability-and-Bioequivalence-Studies-71-320.jpg)



![Maintenance of Records & Retention of
Study Samples
All Records of in vivo tests on any marketed batch of a
drug product should be maintained by the Sponsor
for atleast 2 years after expiry date of the batch
All Drug samples to be retained for a period of atleast
3 years after conduct of the study
OR
1year after expiry of the batch
[Stored in conditions consistent with the product labeling]](https://image.slidesharecdn.com/kunalseminar-bioavailabilityandbioequivalence-130618125726-phpapp01/85/Bioavailability-and-Bioequivalence-Studies-75-320.jpg)




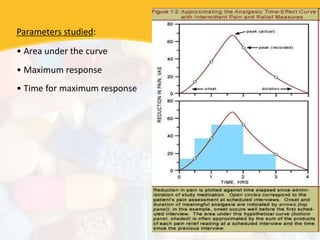











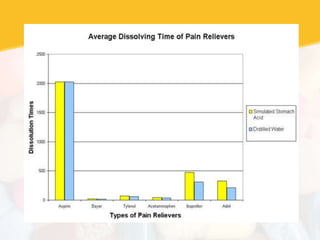




Bioavailability and bioequivalence studies are essential to ensure uniform quality, efficacy, and safety of pharmaceutical products. Bioavailability measures the rate and amount of drug that reaches systemic circulation, while bioequivalence demonstrates that generic and brand name products have comparable rates and extents of absorption. Well-designed pharmacokinetic studies are commonly used to assess bioequivalence by comparing AUC and Cmax of test and reference products. Factors like dosage form, solubility, transit time and metabolism can influence bioavailability, so studies may be necessary after manufacturing changes or for different routes of administration. Guidelines regulate bioequivalence testing to allow approval of lower-cost generic drugs while maintaining therapeutic equivalence.















![Bio pharmaceutical classification System [BCS]](https://cdn.slidesharecdn.com/ss_thumbnails/biopharmaceuticalclassificationystembcs-160328061345-thumbnail.jpg?width=640&height=640&fit=bounds)







































































