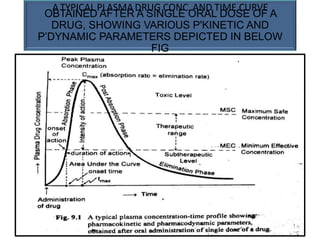
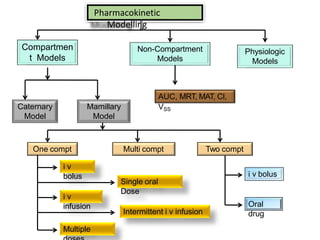
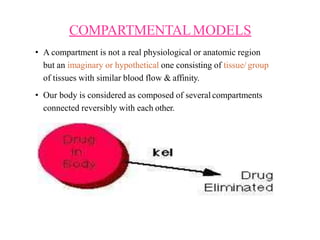
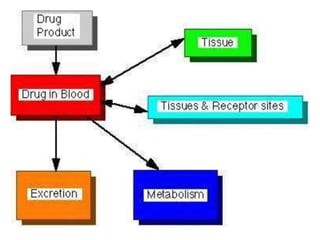
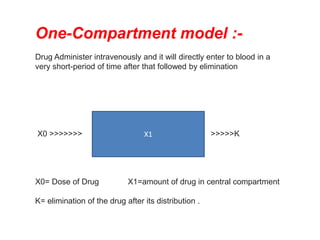
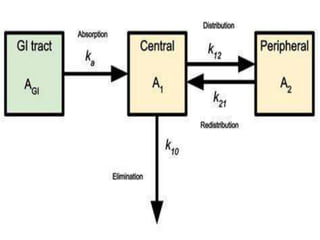
This document provides an overview of pharmacokinetic models and parameters. It discusses basic pharmacokinetic concepts like compartments, absorption, distribution, elimination and clearance. It describes one-compartment and two-compartment models and compares compartmental vs non-compartmental approaches. Key pharmacokinetic parameters like Cmax, Tmax, AUC, volume of distribution, half-life and clearance are also summarized. Organ clearance concepts and the relationship between hepatic blood flow and extraction ratio are covered as well.





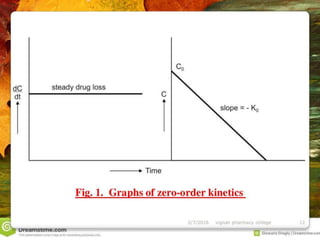
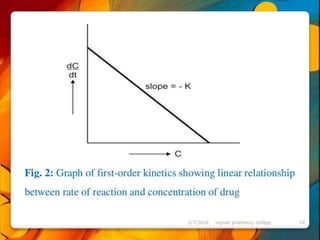

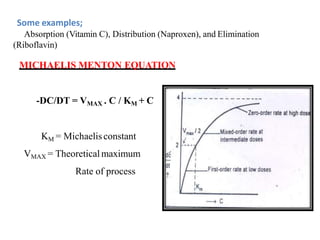
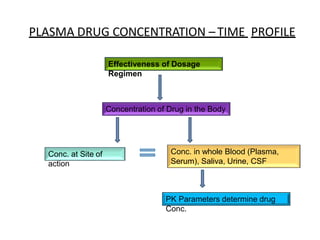















































![industerial 11[r5575677676756767671].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/industerial111-250310082558-cc51ed29-thumbnail.jpg?width=640&height=640&fit=bounds)
![Lab_four__Nausea_and_Vomiting[1].pp powerx](https://cdn.slidesharecdn.com/ss_thumbnails/lab4nauseaandvomiting1-250114210006-ca029c9c-thumbnail.jpg?width=640&height=640&fit=bounds)
![practical lecture. Antihistamine[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/lab3-250124181034-64fb9ab8-thumbnail.jpg?width=640&height=640&fit=bounds)



![3- lab Skin conditions and diseases [1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/3-labskincondition1-250426235724-86d8d5a1-thumbnail.jpg?width=640&height=640&fit=bounds)































